Journal Pre-proof - Genetics in Medicine
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

Journal Pre-proof A Solve-RD ClinVar-based reanalysis of 1,522 index cases from ERN-ITHACA reveals common pitfalls and misinterpretations in exome sequencing Anne-Sophie Denommé-Pichon, Leslie Matalonga, Elke de Boer, Adam Jackson, Elisa Benetti, Siddharth Banka, Ange-Line Bruel, Andrea Ciolfi, Jill Clayton-Smith, Bruno Dallapiccola, Yannis Duffourd, Kornelia Ellwanger, Chiara Fallerini, Christian Gilissen, Holm Graessner, Tobias B. Haack, Marketa Havlovicova, Alexander Hoischen, Nolwenn Jean-Marçais, Tjitske Kleefstra, Estrella López-Martín, Milan Macek, Jr., Maria Antonietta Mencarelli, Sébastien Moutton, Rolph Pfundt, Simone Pizzi, Manuel Posada, Francesca Clementina Radio, Alessandra Renieri, Caroline Rooryck, Lukas Ryba, Hana Safraou, Martin Schwarz, Marco Tartaglia, Christel Thauvin-Robinet, Julien Thevenon, Frédéric Tran Mau-Them, Aurélien Trimouille, Pavel Votypka, Bert B.A. de Vries, Marjolein H. Willemsen, Birte Zurek, Alain Verloes, Christophe Philippe, Solve-RD DITF-ITHACA, Solve-RD SNV-indel working group, Solve-RD Consortia, Orphanomix Group, Antonio Vitobello, Lisenka E.L.M. Vissers, Laurence Faivre PII: S1098-3600(23)00024-2 DOI: https://doi.org/10.1016/j.gim.2023.100018 Reference: GIM 100018 To appear in: Genetics in Medicine Received Date: 14 March 2022 Revised Date: 12 January 2023 Accepted Date: 12 January 2023 Please cite this article as: Denommé-Pichon AS, Matalonga L, de Boer E, Jackson A, Benetti E, Banka S, Bruel AL, Ciolfi A, Clayton-Smith J, Dallapiccola B, Duffourd Y, Ellwanger K, Fallerini C, Gilissen C, Graessner H, Haack TB, Havlovicova M, Hoischen A, Jean-Marçais N, Kleefstra T, López-Martín E, Macek M Jr., Mencarelli MA, Moutton S, Pfundt R, Pizzi S, Posada M, Radio FC, Renieri A, Rooryck C, Ryba L, Safraou H, Schwarz M, Tartaglia M, Thauvin-Robinet C, Thevenon J, Mau-Them FT, Trimouille A, Votypka P, de Vries BBA, Willemsen MH, Zurek B, Verloes A, Philippe C, Solve-RD DITF-ITHACA, Solve-RD SNV-indel working group, Solve-RD Consortia, Orphanomix Group, Vitobello A, Vissers LELM, Faivre L, A Solve-RD ClinVar-based reanalysis of 1,522 index cases from ERN-ITHACA reveals

common pitfalls and misinterpretations in exome sequencing, Genetics in Medicine (2023), doi: https:// doi.org/10.1016/j.gim.2023.100018. This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. © 2023 Published by Elsevier Inc. on behalf of American College of Medical Genetics and Genomics.
A Solve-RD ClinVar-based reanalysis of 1,522
index cases from ERN-ITHACA reveals
common pitfalls and misinterpretations in
exome sequencing
Anne-Sophie Denommé-Pichon1,2, Leslie Matalonga3, Elke de Boer4,5, Adam Jackson6, Elisa Benetti7,
Siddharth Banka6, Ange-Line Bruel1,2, Andrea Ciolfi8, Jill Clayton-Smith6, Bruno Dallapiccola8, Yannis
Duffourd1,2, Kornelia Ellwanger9,10, Chiara Fallerini7,16, Christian Gilissen4,11, Holm Graessner9,10,
Tobias B. Haack9,10, Marketa Havlovicova12, Alexander Hoischen4,11,13, Nolwenn Jean-Marçais2,19,
Tjitske Kleefstra4,5,22, Estrella López-Martín15, Milan Macek Jr.12, Maria Antonietta Mencarelli17,
Sébastien Moutton2, Rolph Pfundt4,5, Simone Pizzi8, Manuel Posada15, Francesca Clementina Radio8,
Alessandra Renieri7,16,17, Caroline Rooryck18, Lukas Ryba12, Hana Safraou1,2, Martin Schwarz12, Marco
Tartaglia8, Christel Thauvin-Robinet1,2,19, Julien Thevenon2, Frédéric Tran Mau-Them1,2, Aurélien
Trimouille20, Pavel Votypka12, Bert B.A. de Vries4,5, Marjolein H. Willemsen4,5, Birte Zurek9,10, Alain
Verloes14,21, Christophe Philippe1,2, Solve-RD DITF-ITHACA, Solve-RD SNV-indel working group, Solve-
RD Consortia, Orphanomix Group, Antonio Vitobello1,2, Lisenka E.L.M. Vissers4,5, Laurence Faivre2,19
1
Unité Fonctionnelle d’Innovation diagnostique des maladies rares, FHU-TRANSLAD, CHU Dijon
Bourgogne, Dijon, France
2
UFR Des Sciences de Santé, INSERM-Université de Bourgogne UMR1231 GAD “Génétique des
Anomalies du Développement”, FHU-TRANSLAD, Dijon, France
3
CNAG‐CRG, Centre for Genomic Regulation”, The Barcelona Institute of Science and Technology,
Barcelona, Spain
4
Dept of Human Genetics, Radboudumc, Nijmegen, The Netherlands
5
Donders Institute for Brain, Cognition and Behaviour, Radboudumc, Nijmegen, The Netherlands
6
Manchester Centre for Genomic Medicine, University of Manchester, Manchester, UK
7
Med Biotech Hub and Competence Center, Dept of Medical Biotechnologies, University of Siena,
Italy
8
Genetics and Rare Diseases Research Division, Ospedale Pediatrico Bambino Gesù, IRCCS, Rome,
Italy
9
Institute of Medical Genetics and Applied Genomics, University of Tübingen, Tübingen, Germany
10
Centre for Rare Diseases, University of Tübingen, Tübingen, Germany
11
Radboud Institute for Molecular Life Sciences, Nijmegen, the Netherlands
12
Dept of Biology and Medical Genetics, Charles University Prague, Prague, Czech Republic
13
Dept of Internal Medicine and Radboud Center for Infectious Diseases (RCI), Radboudumc,
Nijmegen, the Netherlands
14
Dept of Genetics, Assistance Publique-Hôpitaux de Paris - Université de Paris, Paris, France
15
Institute of Rare Diseases Research, Spanish Undiagnosed Rare Diseases Cases Program (SpainUDP)
& Undiagnosed Diseases Network International (UDNI), Instituto de Salud Carlos III, Madrid, Spain
16
Medical Genetics, University of Siena, Siena, Italy
17
Genetica Medica, Azienda Ospedaliero-Universitaria Senese, Siena, Italy
18
University Bordeaux, MRGM INSERM U1211, CHU de Bordeaux, Service de Génétique Médicale,
Bordeaux, France
1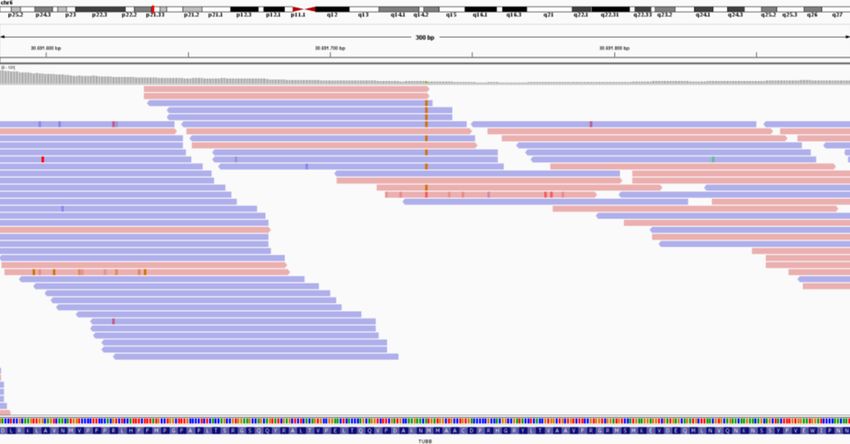
19
Dept of Genetics and Reference Center for Development disorders and intellectual disabilities, FHU
TRANSLAD and GIMI Institute, CHU Dijon Bourgogne, Dijon, France
20
Laboratoire de Génétique Moléculaire, Service de Génétique Médicale, CHU Bordeaux – Hôpital
Pellegrin, Bordeaux, France
21
INSERM UMR 1141 “NeuroDiderot”, Hôpital Robert Debré, Paris, France
22
Center of Excellence for Neuropsychiatry, Vincent van Gogh Institute for Psychiatry, Venray, The
Netherlands
Corresponding authors
Dr Anne-Sophie Denommé-Pichon, MD
Unité Fonctionnelle Innovation en Diagnostic génomique des maladies rares, FHU-TRANSLAD, CHU
Dijon Bourgogne, 15 boulevard Maréchal de Lattre de Tassigny, 21070 Dijon, France
Phone: +33 (0)3 80 39 32 38 / Fax: +33 (0)3 80 29 32 66
E-mail: anne-sophie.denomme-pichon@u-bourgogne.fr
Pr. Laurence Faivre, MD-PhD
FHU-TRANSLAD, Centre de Génétique, Hôpital d’Enfants, CHU Dijon Bourgogne, 14 rue Paul Gaffarel,
21073 Dijon, France
Phone: +33 (0)3 80 29 53 13 / Fax: +33 (0)3 80 29 32 66
E-mail: laurence.faivre@chu-dijon.fr
Keywords: ClinVar, exome reanalysis, rare diseases, developmental disorder
2Abstract (200/200 words)
Purpose
Within the Solve-RD project (https://solve-rd.eu/), the ERN-ITHACA (European Reference
Network for Intellectual disability, TeleHealth, Autism and Congenital Anomalies) aimed to
investigate whether a reanalysis of exomes from unsolved cases based on ClinVar
annotations could establish additional diagnoses. We present the results of the “ClinVar low-
hanging fruit” reanalysis, reasons for the failure of previous analyses and lessons learned.
Methods
Data from the first 3,576 exomes (1,522 probands and 2,054 relatives) collected from ERN-
ITHACA was reanalyzed by the Solve-RD consortium by evaluating for the presence of
SNV/indel already reported as (likely) pathogenic in ClinVar. Variants were filtered on
frequency, genotype and mode of inheritance and reinterpreted.
Results
We identified causal variants in 59 cases (3.9%), 50 of them also raised by other approaches
and 9 leading to new diagnoses, highlighting interpretation challenges: variants in genes not
known to be involved in human disease at the time of the first analysis, misleading genotypes
or variants undetected by local pipelines (variants in off-target regions, low quality filters,
low allelic balance or high frequency).
Conclusion
The “ClinVar low-hanging fruit” analysis represents an effective, fast and easy approach to
recover causal variants from exome sequencing data, herewith contributing to the reduction
of the diagnostic deadlock.
3Introduction
Exome (ES) and genome sequencing (GS) are gold standard tests for the diagnosis of genetic
developmental anomalies. Wright et al. showed that the exome diagnostic rate in pediatric
population was 42% for intellectual disability (ID) [1]. Overall, in developmental disorders,
ES allows positive results in 25% to 45% of the individuals [1]–[3]. Unfortunately, the
remaining 55% to 75% of the cases are still left without a diagnosis.
Most disease-causing variants are located within the coding part of the genome [4] and
therefore most cases could theoretically be solved by an exome analysis. In practice,
however, the tools and knowledge available are sometimes still insufficient or incomplete at
the time of initial analysis. Reanalysis of existing sequencing data offers the possibility to
increase the diagnostic yield in the light of improved bioinformatics pipelines and updated
literature [5]–[21]. Data reanalysis or reevaluation can increase the diagnostic yield by 22-
89% [8], [12], [16], [21], depending on the date of the initial analysis and on the time before
the reanalysis. The American College of Medical Genetics and Genomics (ACMG) has
published a series of points to consider regarding the reevaluation and reanalysis of genomic
test results at various levels [22]. It recommends variant-level reevaluation and case-level
reanalysis every 2 years. In addition, for cases remaining unsolved, it is useful to keep
updated phenotypic descriptions to improve the specificity of the phenotype, as this can help
to increase the diagnostic yield as well. Teams often use a manual approach for routine
reanalysis, but this is not sustainable at scale, especially for iterative reanalyses with unsolved
cases accumulating over time. The bottleneck here is that these procedures are time-
consuming and require dedicated personnel to be completed.
Solve-RD is a Horizon 2020-supported EU study bringing together more than 300 clinicians,
scientists and patient representatives from 51 sites in over 15 European countries. The project
4aims to establish a diagnosis for genetically undiagnosed individuals. The use of different
standardized and automated bioinformatics pipelines makes systematic reinterpretation less
labor-intensive on a large dataset. As part of the Solve-RD project, it is expected that a
dataset composed of more than 19,000 exomes and genomes of genetically unsolved cases
will be reanalyzed in several batches over 5 years [23]. Data is provided by centers associated
with four core European Reference Networks (ERNs), including the ERN-ITHACA
(Intellectual disability, TeleHealth, Autism and Congenital Anomalies). To facilitate such
large-scale reanalysis, multiple working groups for data analysis have been established, each
focusing on a specific type of variant (e.g., copy number variants (CNVs), mitochondrial
variants (mtDNA), de novo variants and mobile element insertions (MEIs)).
The working group focused on the detection of single nucleotide variants (SNV) and small
insertions and deletions of less than 40 bp (indel) (Solve-RD SNV-indel working group)
initially aims to programmatically identify SNVs or indels already annotated in ClinVar as
pathogenic or likely pathogenic (P/LP) variant: the so-called “low-hanging fruit” variants
[19], [24]. ClinVar is a freely accessible public database aggregating information about
genomic variations and their relationship to diseases. It features more than 1.9 million
recorded submissions [25] and contains in particular variants reported as P/LP by the genetics
community.
We present here the results of this first focused reanalysis, referred to as “ClinVar low-
hanging fruit,” on the first batch of the dataset composed of ERN-ITHACA negative ES, and
we report the causes underlying missed diagnoses at first analysis.
5Methods The steps of the study process are shown in Figure 1. Patient inclusion Clinical data, pedigree structure and their corresponding ES or GS raw data (FASTQ, BAM or CRAM format) of 3,576 datasets (including 1,522 from index cases, with ID or developmental disorder, and 2,054 relatives) have been shared internationally by 7 healthcare partners from ERN-ITHACA through the RD-Connect Genome-Phenome Analysis Platform (GPAP) (https://platform.rd-connect.eu/) between April 2018 and November 2019 as described by Zurek et al. [23]. In compliance with the local ethical guidelines and the Declaration of Helsinki, all individuals (or legal representatives) provided informed consent to participate to the Solve-RD project. ES reanalysis and reinterpretation ES and bioinformatics analyses were performed on platforms as previously described [26]– [28] and are detailed in supplemental material. Raw data were reanalyzed using a centralized, automated analysis and filtering approach developed within the RD-Connect GPAP and in the context of the Solve-RD project [19]. We retained SNVs and indels 1) located in genes associated with ID or neurodevelopmental diseases (NDD), according to a list of clinical and research candidate genes provided by ERN-ITHACA (in supplemental material); 2) having a gnomAD allele frequency
additionally, filtered on gnomAD frequency and internal frequency depending on the mode of
inheritance (Figure 2). Frequency filters allow discarding frequent variants which are not
involved in rare diseases and then avoid having to interpret irrelevant variants, including
variants wrongly reported as being P/LP in ClinVar. The annotated variants were
subsequently returned in February 2020 to the original submitting center that had initially
produced the sequencing data for clinical interpretation.
Variants that were considered to be responsible for the phenotype of cases were visualized in
the Integrative Genomics Viewer (IGV) alongside the exome data of the parents when
available for final visual valuation of quality controls (QC). The ACMG and AMP
(Association for Molecular Pathology) criteria [29] for variant classification were used.
Validation by alternative methods was performed locally in the clinical laboratory that
submitted the sample to Solve-RD.
For each diagnosis achieved through this process, we subsequently attempted to determine
the reason why the variants were not identified or discarded at the first analysis. Clinical
laboratories, in parallel to submission of the genetically unsolved cases to Solve-RD, also
performed local reanalysis. Only information about diagnoses obtained through this
reanalysis strategy has to be recorded by the inclusion centers.
7Results
We identified 1,618 SNVs and indels reported as being pathogenic or likely pathogenic in
ClinVar (Table 1, Figure 2) in 980 index cases from 649 trios, 11 duos and 320 singletons.
We identified 147 candidate variants in 127 individuals. Of these, the variant led to a
conclusive molecular diagnosis in 59 individuals (3.9%). Among these 59 cases, 50 (3.3%)
were also solved through local reanalyses in parallel to the Solve-RD project between the
data upload and the reanalysis, in diagnostic or research laboratories, whereas the remaining
9 (0.6%) were solely identified using the Solve-RD infrastructure. For a variety of reasons,
the 88 remaining variants did not solve the cases: single heterozygous variant for autosomal
recessive disorders, no phenotypic fit combined with poor evidence of the pathogenicity
based on the ACMG/AMP criteria about the variants which should not have been reported in
ClinVar as P/LP, variants already returned to the patient but not fully explanatory, or
incidental findings. These variants were rejected based on expert interpretation in the context
of the observed phenotype. We had no dual diagnoses.
For the 9 cases only identified through the ClinVar Solve-RD analysis (Table 2), we
retrospectively looked back at the original ES data and interpretations to understand why the
variant was not considered at the time of the previous analyses. Two cases were not resolved
because the gene was not yet known to be involved in human disease (cases 1 and 2), three
because the variants had been filtered out during data interpretation (cases 3 to 5) and four
because the variants had not been detected by the in-house pipeline or were filtered out
during the bioinformatics filtering process (cases 6 to 9).
Case 1. TRRAP. No disease-gene association at the time of previous analyses
The first solved case was an 8-year-old boy with intrauterine growth retardation, global
developmental delay, severe hypotonia, facial features (cleft palate, short upper lip),
8cerebellar hypoplasia, polymicrogyria and an arachnoid cyst. The reanalysis identified a
heterozygous missense in TRRAP reported as likely pathogenic in ClinVar, leading to the
diagnosis of autosomal dominant developmental delay with or without dysmorphic facies and
autism (MIM #618454). At the time of the first exome analysis in 2018 using a singleton
strategy, the TRRAP gene was not yet known to be involved in human disease. It has been
published in 2019 [30]. Following the identification of the variant as an excellent candidate
for the condition, family segregation by Sanger sequencing confirmed its de novo origin.
Case 2. NFIA. No disease-gene association at the time of previous analyses
The second solved case was a 12-year-old girl with an infantile-onset global developmental
delay associated with behavioral abnormality, hypotonia and corpus callosum hypoplasia.
Dysmorphological features included epicanthal fold, strabismus, wide nasal base,
brachydactyly and clinodactyly of the 2nd toes, one hemangioma on the inner side of the thigh
and thickened skin. At 4 years and 4 months, she weighed 14.0 kg (-1.3 SD), she was 99.5 cm
(-1.3 SD) and her occipital frontal circumference 79.8 cm (-0.4 SD). The reanalysis identified
a heterozygous missense variant in NFIA reported as likely pathogenic in ClinVar, associated
with autosomal dominant brain malformations with or without urinary tract defects (MIM
#613735) [31]. The variant was not identified at the time of the first exome analysis using a
singleton strategy in 2016, because the NFIA gene was not yet reported in OMIM as a morbid
gene. It was associated with human disease in OMIM in 2017. Family segregation by Sanger
sequencing showed that the variant occurred de novo.
Case 3. PTEN. A variant of uncertain significance reclassified as pathogenic
The third solved case was a 9-year-old boy with speech and language developmental delay,
impaired social interactions and poor eye contact, progressive macrocephaly and short corpus
callosum. His father presented with unilateral convergent strabismus, testicular ectopia,
9excessive sweating and macrocephaly (62.5 cm, > +4 SD). His mother also presented with
macrocephaly (61 cm, +4 SD). He was born at 38 weeks of gestation via a normal delivery
following a normal pregnancy. Head circumference at birth was 35.0 cm (-1.5 SD). At the
age of 3, he weighed 17.0 kg (+0.8 SD) and he was 90.0 cm tall (-2.5 SD) and he later
developed macrocephaly (> +2 SD) and obesity (BMI +3.6 SD). He had dysmorphological
features including frontal bossing, telecanthus, esotropia associated with horizontal
nystagmus and one cafe-au-lait spot.
The singleton-based reanalysis identified a heterozygous missense variant in PTEN that had
been previously reported as a variant of uncertain significance (VUS) in the patient because it
was inherited from the mother, who was initially assumed to be unaffected. The variant had
been submitted as pathogenic in ClinVar in the meantime. The Solve-RD reanalysis led us to
reexamine the mother, who was apparently asymptomatic, with mild features compatible with
the diagnosis. This led to the diagnosis of autosomal dominant macrocephaly/autism
syndrome (MIM # 605309), a condition that has been associated with PTEN loss of function.
Case 4. ANO10. Homozygous variant rejected because falsely presumed to be
heterozygous
The fourth solved case is a 41-year-old female with a young adult-onset spinocerebellar
ataxia, associated with nystagmus and hypoplasia of the cerebellar vermis. There was no
medical family history. The first symptoms started at 29 years old with progressive
dysarthria.
The singleton-based reanalysis identified a homozygous indel in ANO10 reported four times
as pathogenic and once as likely pathogenic in ClinVar at the moment of our reanalysis and
already described in several studies [32]. At the time of first analysis, the variant was
examined; however, it was previously wrongly interpreted by the geneticist as heterozygous
10based on inspection of the data in IGV. The genome aligner had produced alignment artifacts,
effectively “hiding” the indel variant due to its position at the end of reads (Figure 3). The
pipeline used HaplotypeCaller and thus did not require realignment around the indels.
Because there is no local realignment around the indels on raw data, the BAM files loaded
into IGV to visualize the variant did not show the indel correctly, suggesting that the
variation was in a heterozygous state.
The new calling was suggestive of a homozygous variant, which we confirmed with Sanger
sequencing verifying the homozygous state, and leading to the diagnosis of autosomal
recessive spinocerebellar ataxia type 10 (MIM #613728).
Case 5. SYNGAP1. Misinterpretation
The fifth solved case was a 12-year-old female with ID, delayed speech and language
development, absence seizures triggered by food intake, autism and behavioral troubles. She
had hyperbilirubinemia during neonatal period and feeding difficulties in infancy. She
presented with hypopigmentation of the skin, recurrent infections, muscular hypotonia, hip
dysplasia and equinovarus deformity.
The reanalysis identified a de novo heterozygous splicing variant in SYNGAP1, reported as
likely pathogenic in ClinVar, leading to the diagnosis of autosomal dominant mental
retardation type 5 (MIM #612621). The variant was missed by the geneticist at the time of the
trio-based analysis (2016), despite SYNGAP1 being a known ID gene at the time of initial
analysis, the variant was reported in the literature [33] and was clearly visible in the BAM
file.
Case 6. TUBB3 Undetected by pipeline
The sixth solved case has been previously described in de Boer et al. [34, p. 3]. This case was
a 16-year-old female with severe neurodevelopmental delay, severe ID and progressive
11microcephaly (-2 SD at 1 year). Family history was negative for genetic diseases and ID. She
presented with delayed motor and communication development and behavioral problems
(auto- and hetero-aggressive behavior, sleep disturbance and phonophobia). She had mild
dysmorphological features, including round face, deeply set eyes, short philtrum, inverted
nipples, pectus carinatum and short feet. She had other medical problems, including short
stature (-2.5 SD), high hypermetropia, strabismus, recurrent upper respiratory and urinary
tract infections, constipation and impaired pain sensation. Brain MRI at age 2 years and 2
months showed notably corpus callosum hypoplasia, reduced volume of supratentorial white
matter and ventriculomegaly.
The reanalysis identified a heterozygous missense variant in TUBB3, reported as likely
pathogenic in ClinVar. The in-house pipeline in the first trio-based analysis (2014) did not
call the variant, because it was located in a region not targeted by the enrichment kit. Variant
calling in the bioinformatics pipeline was based on this target set ± 200 bases, leaving the
variant undetected. We found it in Solve-RD using genome-wide variant calling on raw
datasets, leading to the diagnosis of autosomal dominant cortical dysplasia, complex, with
other brain malformations type 1 (MIM #614039).
Case 7. TUBB. Rejected by the pipeline based on QC filters
The seventh solved case was a 24-year-old female with global developmental delay (motor,
speech and language development) and cerebral palsy. She had severe cortical visual
impairment associated with megalocornea, hypoplasia of the optic nerve, severe myopia,
unilateral strabismus, horizontal nystagmus and photophobia. Dysmorphological features
included ptosis, wide nasal bridge, bulbous nasal tip, low insertion of columella, wide mouth,
thick lower lip vermilion, long fingers with prominent fingertip pads and short feet with pes
planus and hallux valgus. Other medical problems included constipation and Hirschsprung
12disease. Brain MRI showed corpus callosum agenesis, gray matter heterotopias and
abnormality of the caudate nucleus and putamen. Mother presented with (familial occurring)
bilateral coloboma of the iris and retina.
The reanalysis identified a heterozygous pathogenic missense in TUBB reported in the
literature in 2012 [35]. The variant had not passed at initial trio-based analysis in 2013
because of the overall low quality of the variant, likely due to low depth of coverage (23 X
coverage, with 8 variant reads) (Figure 3). Indeed, the in-house pipeline generates a QC score
per variant and this variant had a low score, probably because of the low coverage and
because there is one read with a third allele at the locus. The initial local diagnostic analysis
heavily relied on de novo variant calling, which is less accurate for variants with low QC
scores. Therefore, the variant was likely filtered out during the interpretation process. Now,
the variant led to the diagnosis of autosomal dominant cortical dysplasia, complex, with other
brain malformations type 6 (MIM #615771).
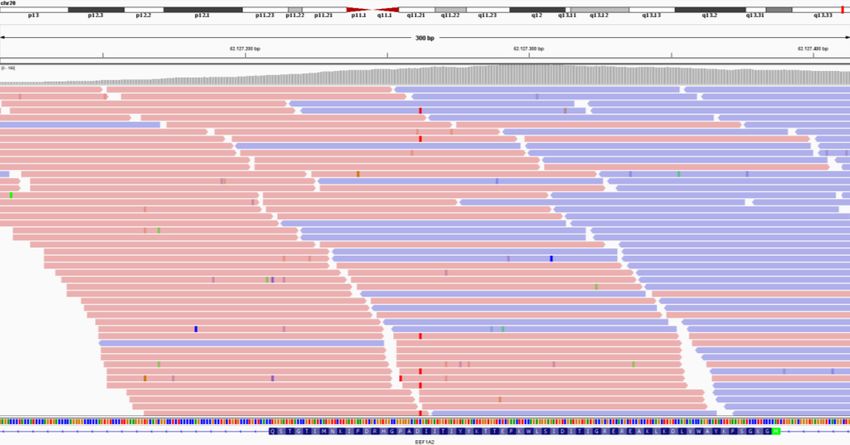
Case 8. EEF1A2. Rejected by pipeline based on allelic balance
The eighth solved case was a 19-year-old male with severe ID associated with global
developmental delay (motor, speech and language development delay), autism (stereotypies,
short attention span, poor eye contact), seizures, hypotonia, sleep disturbance and gait
troubles. Dysmorphological features included macrocephaly, retrognathia, high forehead,
anteverted nares, macrotia, incisor macrodontia, large hands with abnormality of the thumbs
and hip dysplasia. Brain MR imaging showed nonspecific white matter abnormalities around
the left frontal horn.
The reanalysis identified a pathogenic missense variant in EEF1A2, in a mosaic state in the
blood (17%). At the time of the first trio-based analysis in 2014, the position of this variant
was properly covered and the variant was identified with substandard QC, but was
13presumably subsequently rejected based on the allelic fraction being below the threshold for interpretation (
Discussion
The aim of Solve-RD is to solve the diagnostic dead-end via reanalysis, but also to identify
new genes and new molecular mechanisms through data sharing and patient inclusion at a
pan-European level. The purpose of the “ClinVar low-hanging fruit” analysis from 3,576
individuals (including 1,522 index cases) from the ERN-ITHACA cohort was not to identify
new genes but to decrease the diagnostic dead-end. With 59 diagnoses (3.9%), it shows the
usefulness of a systematic reanalysis of negative exomes focused on SNVs and indels
reported as being pathogenic and likely pathogenic in ClinVar, with systematic realignment,
recalling and reinterpretation using up-to-date bioinformatics pipelines [19]. Between the
inclusion of the cases in the Solve-RD project in 2018 and the reanalysis in 2020, 50
candidate cases from the “ClinVar low-hanging fruit” reanalysis were solved in parallel to the
Solve-RD project, mainly through co-occurring reanalyses or reevaluation of the patients (as
individuals without a genetic diagnosis are often advised to recontact their clinician for such
analysis). Nine additional cases (0.6%) were not yet identified locally either because they had
not recontacted their clinician for reanalysis, or if they had, because the variant escaped
detection at any stage of the ES process (enrichment, efficient sequencing, alignment, calling,
annotation, and/or interpretation). These results match those of the previous similar study on
the DDD cohort (13,462 probands) in which Wright et al. were able to identify 112 variants
in 107 probands (0.8%) as possible diagnoses [37].
After collection of the raw data and completion of their bioinformatics reanalysis, the
reinterpretation of the 1,522 negative cases was easy to implement and fast to interpret. There
were only 147 variants after overlap with a disease-specific gene list and prioritization.
Whereas this list limited the possibility to identify so-called “unanticipated diagnosis,”
referring to phenocopies of a genetical different disease group (e.g. ID genes vs. epilepsy
15genes), this approach does help to limit the risks of incidental findings. In addition, the in
silico enrichment filters also limited the needs in human resources, as the centers did not have
to filter the thousands of cases manually. Moreover, the cohort analysis with only candidate
variants to reevaluate resulted in timesaving compared to a case-by-case reanalysis
considering different strategies.
Our centralized reanalysis has taught us some lessons. The first and very essential one is that
stringent filters should not be used for variants reported as pathogenic or likely pathogenic in
ClinVar: 1) if there is a variant with low depth of coverage: consider it nonetheless for the
interpretation, 2) if there is a variant with low allelic balance: consider mosaicism, 3) if there
is an inherited variant: check for paucisymptomatic parent or consider incomplete penetrance,
4) if there is a frequent variant: check for recurrent variants and involvement in recessive
disorder and use homozygous count annotation in gnomAD. The second lesson is to remain
cautious when interpreting variants located at the end of the reads, especially in IGV.
Genome aligners can produce alignment artifacts and hide indels located at the end of the
reads, especially when there was no realignment around the indel step in the bioinformatic
pipeline, as it can be the case when using UnifiedGenotyper. In these cases, the variant
calling is correct, but the BAM still contains the misalignment. Even though most current
pipelines use variant callers with a reassembly step, like HaplotypeCaller or Platypus, and
thus do not require local indel realignment, it is still useful for legacy tools like
UnifiedGenotyper to correct mapping errors made by genome aligners [38], [39]. We have to
keep in mind that when looking at variations in IGV located at the end of reads, there may be
artifacts related to the fact that there was no realignment around the indels. The third lesson is
to never stop reanalyzing genomic data because new genes and new variants are reported all
the time. Some tools allow automating the reanalysis in real time [13], such as Variant Alert
16[40], which can highlight updates from ClinVar as soon as variants are submitted at the scale
of the complete database.
ClinVar-based reanalysis is an effective technique for solving unsolved cases from the ERN-
ITHACA cohort with intellectual disability and congenital abnormalities and this strategy
should also be effective for other types of cohorts such as those of Solve-RD project. Given
the success of the ClinVar-based reanalysis in diagnosing unsolved cases, similar strategies
for other variant types, like CNVs, may be equally successful, which are currently ongoing.
This approach should be used as part of a more comprehensive reanalysis strategy, since it
only targets variants already reported and cannot resolve all cases. Other strategies should be
considered in reanalysis, like the identification of de novo variants from trio, CNVs,
mitochondrial variants, mobile element insertions, short tandem repeat expansions,
uniparental disomy and variants in runs of homozygosity. However, ES reanalysis alone will
not solve the other causes of diagnostic impasse and additional experiments like GS or
multiomic analyses will be therefore required.
Data availability
Genomic and phenotypic data used during the current study are available on the RD-Connect
GPAP platform, accessible to researchers and clinicians (https://platform.rd-
connect.eu/userregistration/). The RD-Connect identifiers of the cases of this cohort are
available from the corresponding author on request.
17Acknowledgments
We are very grateful to the families participating in the Solve-RD project. We thank the
computer cluster of the University of Burgundy for technical support and management of the
Dijon-Bourgogne computing platform.
Funding
The Solve-RD project has received funding from the European Union’s Horizon 2020
research and innovation program under grant agreement number 779257. Data was analyzed
using the RD-Connect Genome‐ Phenome Analysis Platform, which received funding from
EU projects RD‐ Connect, Solve-RD and EJP-RD (grant numbers FP7 305444, H2020
779257, H2020 825575), Instituto de Salud Carlos III (grant numbers PT13/0001/0044,
PT17/0009/0019; Instituto Nacional de Bioinformática, INB) and ELIXIR Implementation
Studies. The collaborations in this study were facilitated by ERN-ITHACA, one of the 24
European Reference Networks (ERNs) approved by the ERN Board of Member States, co-
funded by the European Commission. This project was supported by CZ MZCR.cz n.
00064203 and by MSMT.cz n. - LM2018132 to MM.
Author Contributions
Data curation: A.S.D.P.; E.d.B.; A.C.; N.J.M; S.M.; S.P.; F.C.R.: M.S.; M.T. Formal
analysis: A.S.D.P.; E.d.B.; T.B.H.; C.T.; F.T.M.T.; C.P. Funding acquisition: H.G.: B.Z.
Investigation: A.S.D.P.; A.T. Project administration: K.E.; H.G.; B.Z. Software: L.M.; A.H.
Supervision: B.D.; M.T.; L.E.L.M.V.; L.F. Writing-original draft: A.S.D.P. Writing-review &
editing: A.S.D.P.; L.M.; E.d.B.; E.B.; A.C.; B.D.; C.F.; C.G.; H.G.; T.B.H.; E.L.M.; M.A.M.;
S.P.; M.P.; F.C.R.; A.R.; M.T.; C.T.; F.T.M.T.; B.B.A.d.V.; C.P.; A.V.; L.E.L.M.V.; L.F.
18Ethical approval
This study was approved by the appropriate French independent ethics committee (Comité de
Protection des Personnes Ouest I 2019T2-12). Individuals from Prague were included in the
collection from the Coordinating Center for Patients with Rare Diseases (number NCCVO
135V32R001102, 21.07.2015). Individuals from Nijmegen were included in the Solve-RD
collection (ID: 2018-4986, 28.11.2018; Biobank contract between department and
Radboudumc BiobankID: 2014-1254, 2014-09-09). Individuals from Roma were included
using the European Commission’s information system for patient sharing as part of what is
provided for European Reference Networks for Rare Diseases (2017-11-22). Individuals from
Siena were included in the NeuroNext collection (Identificazione di geni coinvolti nella
patogenesi dei disturbi del neurosviluppo e delle malattie neurodegenerative, 2020-05-18).
Individuals from Madrid provided informed consent for the donation of biological samples to
the National Rare Diseases Biobank of the Instituto de Investigación de enfermedades raras
V.1 (ID: CEI PI 74_2016, 2017-02-06).
Disclosure
The authors declare no conflict of interest. The Department of Molecular and Human
Genetics at Baylor College of Medicine receives revenue from clinical genetic testing
completed at Baylor Genetics Laboratories.
Solve-RD DITF-ITHACA
Kristin M. Abbott[1], Siddharth Banka[2, 3], Elke de Boer[4, 5], Andrea Ciolfi[6], Jill Clayton-
Smith[2, 3], Bruno Dallapiccola[6], Anne-Sophie Denommé-Pichon[7], Laurence Faivre[7-11],
Christian Gilissen[4, 12], Tobias B. Haack[13], Marketa Havlovicova[14], Alexander Hoischen[4,
1912, 15]
, Adam Jackson[2, 3], Mieke Kerstjens[1], Tjitske Kleefstra[4, 5], Estrella López Martín[16],
Milan Macek Jr.[14], Leslie Matalonga[17], Isabelle Maystadt[18], Manuela Morleo[19], Vicenzo
Nigro[19, 20], Michele Pinelli[19], Simone Pizzi[6], Manuel Posada[16], Francesca C. Radio[6],
Alessandra Renieri[21-23], Olaf Riess[13, 24], Caroline Rooryck[25, 26], Lukas Ryba[14], Jean-
Madeleine de Sainte Agathe[27], Gijs W.E. Santen[28], Martin Schwarz[14], Marco Tartaglia[6],
Christel Thauvin[7-11], Annalaura Torella[20], Aurélien Trimouille[25, 26], Alain Verloes[29, 30],
Lisenka Vissers[4, 5], Antonio Vitobello[7], Pavel Votypka[14], and Kristina Zguro[22].
1. Department of Genetics, University Medical Center Groningen, University of Groningen,
Groningen, The Netherlands.
2. Division of Evolution, Infection and Genomics, School of Biological Sciences, Faculty of
Biology, Medicine and Health, University of Manchester, Manchester M13 9WL, UK.
3. Manchester Centre for Genomic Medicine, St Mary’s Hospital, Manchester University
Hospitals NHS Foundation Trust, Health Innovation Manchester, Manchester M13 9WL,
UK.
4. Department of Human Genetics, Radboud University Medical Center, Nijmegen, the
Netherlands.
5. Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical
Center, Nijmegen, the Netherlands.
6. Genetics and Rare Diseases Research Division, Ospedale Pediatrico Bambino Gesù,
IRCCS, 00146 Rome, Italy.
7. Inserm - University of Burgundy-Franche Comté, UMR1231 GAD, Dijon, France.
8. Dijon University Hospital, Centre of Reference for Rare Diseases: Development disorders
and malformation syndromes, Dijon, France.
9. Dijon University Hospital, FHU-TRANSLAD, Dijon, France.
10. Dijon University Hospital, Genetics Department, Dijon, France.
11. Dijon University Hospital, GIMI institute, Dijon, France.
12. Radboud Institute for Molecular Life Sciences, Nijmegen, The Netherlands.
13. Institute of Medical Genetics and Applied Genomics, University of Tübingen, Tübingen,
Germany.
14. Department of Biology and Medical Genetics, Charles University Prague-2nd Faculty of
Medicine and University Hospital Motol, Prague, Czech Republic.
15. Department of Internal Medicine and Radboud Center for Infectious Diseases (RCI),
Radboud University Medical Center, Nijmegen, the Netherlands.
16. Institute of Rare Diseases Research, Spanish Undiagnosed Rare Diseases Cases Program
(SpainUDP) & Undiagnosed Diseases Network International (UDNI), Instituto de Salud
Carlos III, Madrid, Spain.
17. CNAG-CRG, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science
and Technology, Baldiri Reixac 4, Barcelona 08028, Spain.
2018. Centre de Génétique Humaine, Institut de Pathologie et de Génétique, Gosselies,
Belgium.
19. Telethon Institute of Genetics and Medicine, Pozzuoli, Italy.
20. Dipartimento di Medicina di Precisione, Università degli Studi della Campania "Luigi
Vanvitelli," Napoli, Italy.
21. Genetica Medica, Azienda Ospedaliero-Universitaria Senese, Italy.
22. Med Biotech Hub and Competence Center, Department of Medical Biotechnologies,
University of Siena, Italy.
23. Medical Genetics, University of Siena, Italy.
24. Centre for Rare Diseases, University of Tübingen, Tübingen, Germany.
25. MRGM, Maladies Rares: Génétique et Métabolisme, lNSERM U1211, Université de
Bordeaux, Bordeaux, France.
26. Service de Génétique Médicale, Centre Hospitalier Universitaire de Bordeaux, Bordeaux,
France.
27. Department of genetics, Assistance Publique-Hôpitaux de Paris - Sorbonne Université,
Pitié-Salpêtrière University Hospital, 83 Boulevard de l’Hôpital, Paris, France.
28. Department of Clinical Genetics, Leiden University Medical Center, Leiden, The
Netherlands.
29. Département de Génétique, Hôpital Robert DEBRE, 48 boulevard Serurier, 75019 Paris,
France.
30. INSERM UMR 1141 "NeuroDiderot", Hôpital Robert DEBRE, Paris, France.
Solve-RD SNV-indel working group
Elke de Boer[1, 2], Enzo Cohen[3], Daniel Danis[4], Anne-Sophie Denommé-Pichon[5], Fei
Gao[6], Christian Gilissen[1, 7], Rita Horvath[6], Mridul Johari[8], Lennart Johanson[9], Shuang
Li[9], Leslie Matalonga[10], Heba Morsy[11], Isabelle Nelson[3], Ida Paramonov[10], Iris B.A.W.
te Paske[1, 7], Peter Robinson[4], Marco Savarese[8], Wouter Steyaert[1, 7], Ana Töpf[12],
Aurélien Trimouille[13, 14], Joeri K. van der Velde[9], Jana Vandrovcova[11], Antonio
Vitobello[5].
1. Department of Human Genetics, Radboud University Medical Center, Nijmegen, the
Netherlands.
2. Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical
Center, Nijmegen, the Netherlands.
3. Sorbonne Université, Inserm, Institut de Myologie, Centre de Recherche en Myologie, F-
75013 Paris, France.
4. Jackson Laboratory for Genomic Medicine, Farmington, CT 06032, USA.
5. Inserm - University of Burgundy-Franche Comté, UMR1231 GAD, Dijon, France.
216. Department of Clinical Neurosciences, School of Clinical Medicine, John Van Geest
Centre for Brain Repair, University of Cambridge, UK.
7. Radboud Institute for Molecular Life Sciences, Nijmegen, The Netherlands.
8. Folkhälsan Research Center, University of Helsinki, Finland.
9. Department of Genetics, University Medical Center Groningen, University of Groningen,
Groningen, The Netherlands.
10. CNAG-CRG, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science
and Technology, Baldiri Reixac 4, Barcelona 08028, Spain.
11. Department of Neuromuscular Diseases, UCL Queen Square Institute of Neurology,
London, UK.
12. John Walton Muscular Dystrophy Research Centre, Translational and Clinical Research
Institute, Newcastle University and Newcastle Hospitals NHS Foundation Trust, Newcastle
upon Tyne, UK.
13. MRGM, Maladies Rares: Génétique et Métabolisme, lNSERM U1211, Université de
Bordeaux, Bordeaux, France.
14. Service de Génétique Médicale, Centre Hospitalier Universitaire de Bordeaux, Bordeaux,
France.
Solve-RD Consortia
EKUT: Olaf Riess1,2, Tobias B. Haack1, Holm Graessner1,2, Birte Zurek1,2,Kornelia
Ellwanger1,2, Stephan Ossowski1,3, German Demidov1, Marc Sturm1, Julia M. Schulze-
Hentrich1, Rebecca Schüle4,5, Jishu Xu4,5, Christoph Kessler4,5, Melanie Wayand4,5, Matthis
Synofzik4,5, Carlo Wilke4,5, Andreas Traschütz4,5, Ludger Schöls4,5, Holger Hengel4,5, Holger
Lerche6, Josua Kegele6, Peter Heutink4,5
RUMC: Han Brunner7,8,9, Hans Scheffer7,8, Nicoline Hoogerbrugge7,10, Alexander
Hoischen7,10,11, Peter A.C. ‘t Hoen,10,12, Lisenka E.L.M. Vissers7, 9, Christian Gilissen7,10,
Wouter Steyaert7,10, Karolis Sablauskas7, Richarda M. de Voer7,10, Erik-Jan Kamsteeg7, Bart
van de Warrenburg9,13, Nienke van Os9,13, Iris te Paske7,10, Erik Janssen7,10, Elke de Boer7, 9,
Marloes Steehouwer7, Burcu Yaldiz7, Tjitske Kleefstra7, 9
University of Leicester: Anthony J. Brookes14, Colin Veal14, Spencer Gibson14, Vatsalya
Maddi14, Mehdi Mehtarizadeh14, Umar Riaz14, Greg Warren14, Farid Yavari Dizjikan14,
Thomas Shorter14,
UNEW: Ana Töpf15, Volker Straub15, Chiara Marini Bettolo15, Jordi Diaz Manera15, Sophie
Hambleton16, Karin Engelhardt17
MUH: Jill Clayton-Smith18,19, Siddharth Banka18,19, Elizabeth Alexander19, Adam
Jackson18,19
DIJON: Laurence Faivre20,21,22,23,24, Christel Thauvin20,21,22,23,24, Antonio Vitobello22, Anne-
Sophie Denommé-Pichon22, Yannis Duffourd22,23, Ange-Line Bruel22, Christine Peyron25,26,
Aurore Pélissier25,26
CNAG-CRG: Sergi Beltran27,28, Ivo Glynne Gut27,28, Steven Laurie27, Davide Piscia27, Leslie
Matalonga27, Anastasios Papakonstantinou27, Gemma Bullich27, Alberto Corvo27, Marcos
Fernandez-Callejo27, Carles Hernández27, Daniel Picó27, Ida Paramonov27, Hanns
Lochmüller27
22EURORDIS: Gulcin Gumus29, Virginie Bros-Facer30
INSERM-Orphanet: Ana Rath31, Marc Hanauer31, David Lagorce31,Oscar
Hongnat31,Maroua Chahdil31,Emeline Lebreton31
INSERM-ICM: Giovanni Stevanin32,33,34,35,36, Alexandra Durr32.33.34.35.37, Claire-Sophie
Davoine32.33.34.35.36, Léna Guillot-Noel32.33.34.35.36, Anna Heinzmann32.33.34.35.38, Giulia
Coarelli32.33.34.35.38
INSERM-CRM: Gisèle Bonne39, Teresinha Evangelista39, Valérie Allamand39, Isabelle
Nelson39, Rabah Ben Yaou39,40,41, Corinne Metay39,42, Bruno Eymard39,40, Enzo Cohen39,
Antonio Atalaia39, Tanya Stojkovic39,40
Univerzita Karlova: Milan Macek Jr.43, Marek Turnovec43, Dana Thomasová43, Radka
Pourová Kremliková43, Vera Franková43, Markéta Havlovicová 43, Petra Lišková44,45, Pavla
Doležalová46
EMBL-EBI: Helen Parkinson47, Thomas Keane47, Mallory Freeberg47, Coline Thomas47,
Dylan Spalding47
Jackson Laboratory: Peter Robinson48, Daniel Danis48
KCL: Glenn Robert49, Alessia Costa50, Christine Patch51,52
UCL-IoN: Mike Hanna53, Henry Houlden54, Mary Reilly53, Jana Vandrovcova54, Stephanie
Efthymiou54, Heba Morsy54, Elisa Cali54, Francesca Magrinelli55, Sanjay M. Sisodiya56,
Jonathan Rohrer57
UCL-ICH, Francesco Muntoni58,59, Irina Zaharieva58, Anna Sarkozy58
Universiteit Antwerpen: Vincent Timmerman60,61, Jonathan Baets62,63, Geert de Vries61,62,
Jonathan De Winter61,62,63, Danique Beijer60,61,62, Peter de Jonghe61,63, Liedewei Van de
Vondel60,61,62, Willem De Ridder61,62,63, Sarah Weckhuysen64,65
Uni Naples/Telethon UDP: Vincenzo Nigro66,67, Margherita Mutarelli67,68, Manuela
Morleo67, Michele Pinelli67, Alessandra Varavallo67, Sandro Banfi66,67, Annalaura Torella66,
Francesco Musacchia66,67, Giulio Piluso66
UNIFE: Alessandra Ferlini69, Rita Selvatici69, Francesca Gualandi69, Stefania Bigoni69,
Rachele Rossi69, Marcella Neri69
UKB: Stefan Aretz70,71, Isabel Spier70,71, Anna Katharina Sommer70, Sophia Peters70
IPATIMUP: Carla Oliveira72,73,74, Jose Garcia Pelaez72,73,75, Ana Rita Matos72,73,76, Celina
São José72,73,75, Marta Ferreira72,73,77, Irene Gullo72,73,78, Susana Fernandes72,79, Luzia
Garrido80, Pedro Ferreira72,73,81, Fátima Carneiro72,73,78
UMCG: Morris A Swertz82, Lennart Johansson82, Joeri K van der Velde82, Gerben van der
Vries82, Pieter B Neerincx82, David Ruvolo82, Kristin M Abbott83, Wilhemina S Kerstjens
Frederikse83,84, Eveline Zonneveld-Huijssoon83,85, Dieuwke Roelofs-Prins82, Marielle van
Gijn83,85
Charité: Sebastian Köhler86
SHU: Alison Metcalfe87,88
APHP: Alain Verloes89,90, Séverine Drunat89,90, Delphine Heron91,92, Cyril Mignot91,93, Boris
Keren91, Jean-Madeleine de Sainte Agathe91
CHU Bordeaux: Caroline Rooryck94, Didier Lacombe94, Aurelien Trimouille95
Spain UDP: Manuel Posada De la Paz96, Eva Bermejo Sánchez96, Estrella López Martín96,
Beatriz Martínez Delgado96, F. Javier Alonso García de la Rosa96
Ospedale Pediatrico Bambino Gesù, Rome: Andrea Ciolfi97, Bruno Dallapiccola97, Simone
Pizzi97, Francesca Clementina Radio97, Marco Tartaglia97
University of Siena: Alessandra Renieri98,99,100, Simone Furini98,99, Chiara Fallerini98,99, Elisa
Benetti98,99
Semmelweis University Budapest: Peter Balicza101, Maria Judit Molnar101
University of Ljubljana, Ales Maver102, Borut Peterlin102
23University of Lübeck: Alexander Münchau103, Katja Lohmann104, Rebecca Herzog103,105,
Martje Pauly103,104
Val d’Hebron Barcelona: Alfons Macaya106,107, Ana Cazurro-Gutiérrez106, Belén Pérez-
Dueñas106, Francina Munell106, Clara Franco Jarava108,109, Laura Batlle Masó110,111, Anna
Marcé-Grau106, Roger Colobran108,109,112
Hospital Sant Joan de Déu Barcelona: Andrés Nascimento Osorio113
, Daniel Natera de Benito113
University of Freiburg: Hanns Lochmüller114,115,116, Rachel Thompson116, Kiran
Polavarapu116, Bodo Grimbacher117,118,119,120,121
University of Oxford: David Beeson122, Judith Cossins122
Folkhälsan Research Centre: Peter Hackman123, Mridul Johari123, Marco Savarese123,
Bjarne Udd123,124,125
University of Cambridge: Rita Horvath126, Patrick F. Chinnery126,127, Thiloka Ratnaike128,
Fei Gao126, Katherine Schon126,129
Catalan Institute of Oncology, Barcelona: Gabriel Capella130, Laura Valle130
KU Munich: Elke Holinski-Feder131, Andreas Laner132, Verena Steinke-Lange131
TU Dresden: Evelin Schröck133, Andreas Rump133,134
Koç University: Ayşe Nazlı Başak135
Ghent University Hospital: Dimitri Hemelsoet136,137, Bart Dermaut137,138,139, Nika
Schuermans137,138,139, Bruce Poppe137,138,139, Hannah Verdin137138
University Hospital Meyer, Florence: Davide Mei140, Annalisa Vetro140, Simona
Balestrini140,141, Renzo Guerrini140
KU Leuven: Kristl Claeys142,143
LUMC: Gijs W.E. Santen144, Emilia K. Bijlsma144, Mariette J.V. Hoffer144, Claudia A.L.
Ruivenkamp144
Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna: Kaan
Boztug145,146,147,148,149, Matthias Haimel145,146,147
Institute of Pathology and Genetics, Gosselies, Belgium: Isabelle Maystadt150,151
Technical University Munich: Isabell Cordts152, Marcus Deschauer152
Neurology/Neurogenetics Laboratory University of Crete, Heraklion, Crete, Greece:
Ioannis Zaganas153, Evgenia Kokosali153, Mathioudakis Lambros153, Athanasios
Evangeliou154, Martha Spilioti155, Elisabeth Kapaki156, Mara Bourbouli156
IRCCS G. Gaslini: Pasquale Striano157,158, Federico Zara158,159, Antonella Riva158,159,
Michele Iacomino159,160, Paolo Uva160, Marcello Scala157,158, Paolo Scudieri158,159
Cliniques universitaires Saint-Luc (CUSL): Maria-Roberta Cilio161, Evelina Carpancea161,
Chantal Depondt162, Damien Lederer163, Yves Sznajer164, Sarah Duerinckx165, Sandrine
Mary163
Institute of Human Genetics, University Hospital Essen: Christel Depienne166,167, Andreas
Roos168,169,170
University of Luxembourg: Patrick May171
1
Institute of Medical Genetics and Applied Genomics, University of Tübingen, Tübingen, Germany.
2
Centre for Rare Diseases, University of Tübingen, Tübingen, Germany.
3
NGS Competence Center Tübingen (NCCT), University of Tübingen, Tübingen, Germany.
244
Department of Neurodegeneration, Hertie Institute for Clinical Brain Research (HIH), University of Tübingen,
Tübingen, Germany.
5
German Center for Neurodegenerative Diseases (DZNE), Tübingen, Germany.
6
Department of Neurolgy and Epileptology, Hertie Institute for Clinical Brain Research (HIH), University of
Tübingen, Tübingen, Germany.
7
Department of Human Genetics, Radboud University Medical Center, Nijmegen, the Netherlands.
8
Department of Clinical Genetics, Maastricht University Medical Centre, Maastricht, the Netherlands.
9
Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Center, Nijmegen, the
Netherlands.
10
Radboud Institute for Molecular Life Sciences, Nijmegen, The Netherlands.
11
Department of Internal Medicine and Radboud Center for Infectious Diseases (RCI), Radboud University
Medical Center, Nijmegen, the Netherlands.
12
Center for Molecular and Biomolecular Informatics, Radboud University Medical Center, Nijmegen, the
Netherlands
13
Department of Neurology, Radboud University Medical Center, Nijmegen, The Netherlands.
14
Department of Genetics and Genome Biology, University of Leicester, Leicester, UK.
15
John Walton Muscular Dystrophy Research Centre, Translational and Clinical Research Institute, Newcastle
University and Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, UK.
16
Primary Immunodeficiency Group, Translational and Clinical Research Institute, Newcastle University and
Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, UK.
17
Primary Immunodeficiency Group, Translational and Clinical Research Institute, Newcastle University,
Newcastle upon Tyne, UK.
18
Division of Evolution, Infection and Genomics, School of Biological Sciences, Faculty of Biology, Medicine
and Health, University of Manchester, Manchester M13 9WL, UK.
19
Manchester Centre for Genomic Medicine, St Mary’s Hospital, Manchester University Hospitals NHS
Foundation Trust, Health Innovation Manchester, Manchester M13 9WL, UK.
20
Dijon University Hospital, Genetics Department, Dijon, France
21
Dijon University Hospital, Centre of Reference for Rare Diseases: Development disorders and malformation
syndromes, Dijon, France
22
Inserm - University of Burgundy-Franche Comté, UMR1231 GAD, Dijon, France
23
Dijon University Hospital, FHU-TRANSLAD, Dijon, France
24
Dijon University Hospital, GIMI institute, Dijon, France
25
University of Burgundy-Franche Comté, Dijon Economics Laboratory, Dijon, France
26
University of Burgundy-Franche Comté, FHU-TRANSLAD, Dijon, France
27
CNAG‐ CRG, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology,
Baldiri Reixac 4, Barcelona 08028, Spain
28
Universitat Pompeu Fabra (UPF), Barcelona, Spain.
29
EURORDIS-Rare Diseases Europe, Sant Antoni Maria Claret 167 - 08025 Barcelona, Spain
30
EURORDIS-Rare Diseases Europe, Plateforme Maladies Rares, 75014 Paris, France
31
INSERM, US14 - Orphanet, Plateforme Maladies Rares, 75014 Paris, France.
32
Institut National de la Santé et de la Recherche Medicale (INSERM) U1127, Paris, France.
33
Centre National de la Recherche Scientifique, Unité Mixte de Recherche (UMR) 7225, Paris, France.
34
Unité Mixte de Recherche en Santé 1127, Université Pierre et Marie Curie (Paris 06), Sorbonne Universités,
Paris, France.
35
Institut du Cerveau -ICM, Paris, France.
36
Ecole Pratique des Hautes Etudes, Paris Sciences et Lettres Research University, Paris, France.
37
Centre de Référence de Neurogénétique, Hôpital de la Pitié-Salpêtrière, Assistance Publique-Hôpitaux de
Paris (AP-HP), Paris, France.
38
Hôpital de la Pitié-Salpêtrière, Assistance Publique-Hôpitaux de Paris (AP-HP), Paris, France.
39
Sorbonne Université, Inserm, Institut de Myologie, Centre de Recherche en Myologie, F-75013 Paris, France
40
AP-HP, Centre de Référence de Pathologie Neuromusculaire Nord, Est, Ile-de-France, Institut de Myologie,
G.H. Pitié-Salpêtrière, F-75013 Paris, France.
41
Institut de Myologie, Equipe Bases de données, G.H. Pitié-Salpêtrière, F-75013 Paris, France.
2542
AP-HP, Unité Fonctionnelle de Cardiogénétique et Myogénétique Moléculaire et Cellulaire, G.H. Pitié-
Salpêtrière, F-75013 Paris, France.
43
Department of Biology and Medical Genetics, Charles University Prague-2nd Faculty of Medicine and
University Hospital Motol, Prague, Czech Republic.
44
Department of Paediatrics and Inherited Metabolic Disorders, First Faculty of Medicine, Charles University
and General University Hospital in Prague, Prague, Czech Republic.
45
Department of Ophthalmology, First Faculty of Medicine, Charles University and General University
Hospital in Prague, Prague, Czech Republic.
46
Centre for Paediatric Rheumatology and Autoinflammatory Diseases, Department of Paediatrics and Inherited
Metabolic Disorders, 1st Faculty of Medicine, Charles University and General University Hospital in Prague,
Czech Republic
47
European Bioinformatics Institute, European Molecular Biology Laboratory, Wellcome Genome Campus,
Hinxton, Cambridge, United Kingdom.
48
Jackson Laboratory for Genomic Medicine, Farmington, CT 06032, USA.
49
Florence Nightingale Faculty of Nursing, Midwifery & Palliative Care, King’s College, London, UK.
50
Wellcome Genome Campus Society and Ethics Research, Wellcome Genome Campus, UK
51
Genomics England, Queen Mary University of London, Dawson Hall, EC1M 6BQ, London, UK.
52
Society and Ethics Research, Connecting Science, Wellcome Genome Campus,
Hinxton, UK
53
MRC Centre for Neuromuscular Diseases and National Hospital for Neurology and Neurosurgery, UCL
Queen Square Institute of Neurology, London, UK.
54
Department of Neuromuscular Diseases, UCL Queen Square Institute of Neurology, London, UK.
55
Department of Clinical and Movement Neurosciences, UCL Queen Square Institute of Neurology, University
College London, WC1N 3BG, UK.
56
Department of Clinical and Experimental Epilepsy, UCL Queen Square Institute of Neurology, London, UK.
57
Dementia Research Centre, Department of Neurodegenerative Disease, UCL Queen Square Institute of
Neurology, London, UK.
58
Dubowitz Neuromuscular Centre, UCL Great Ormond Street Hospital, London, UK.
59
NIHR Great Ormond Street Hospital Biomedical Research Centre, London, United Kingdom.
60
Peripheral Neuropathy Research Group, University of Antwerp, Antwerp, Belgium.
61
Laboratory of Neuromuscular Pathology, Institute Born-Bunge, University of Antwerp, Antwerpen, Belgium
62
Translational Neurosciences, Faculty of Medicine and Health Sciences, University of Antwerp, Belgium
63
Neuromuscular Reference Centre, Department of Neurology, Antwerp University Hospital, Antwerpen,
Belgium
64
Translational Neuroscience group, University of Antwerp, Belgium
65
VIB-CMN, Applied and Translational Neurogenomics Group
66
Dipartimento di Medicina di Precisione, Università degli Studi della Campania "Luigi Vanvitelli," Napoli,
Italy.
67
Telethon Institute of Genetics and Medicine, Pozzuoli, Italy.
68
Istituto di Scienze Applicate e Sistemi Intelligenti "E.Caianiello" - ISASI -CNR
69
Unit of Medical Genetics, Department of Medical Sciences, University of Ferrara, Italy.
70
Institute of Human Genetics, Medical Faculty, University of Bonn, Bonn, Germany.
71
Center for Hereditary Tumor Syndromes, University Hospital Bonn, Bonn, Germany.
72
i3S - Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Portugal.
73
IPATIMUP - Institute of Molecular Pathology and Immunology of the University of Porto, Portugal
74
Departament of Pathology, Faculty of Medicine, University of Porto, Portugal.
75
Doctoral Programme in Biomedicine, Faculty of Medicine, University of Porto, Portugal.
76
Doctoral Programme in BiotechHealth, School of Medicine and Biomedical Sciences, University of Porto,
Portugal
77
Doctoral Programme in Computer Science, Faculty of Sciences, University of Porto, Portugal
78
Departament of Pathology, Faculty of Medicine, University of Porto, Portugal.
79
Departament of Genetics, Faculty of Medicine, University of Porto, Portugal.
80
CHUSJ, Centro Hospitalar e Universitário de São João, Porto, Portugal
81
Faculty of Sciences, University of Porto, Portugal
26You can also read

















































