ESR13: Quantum machine learning for reactivity (WP3)
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

ESR13: Quantum machine learning for reactivity (WP3)

Theoretical Chemical Physics group
Headed by Prof. Dr. Alexandre Tkatchenko
Ø Machine learning in Chemical Physics
Ø Non-covalent interactions
Ø Imaginary time path-integrals
https://www.tcpunilu.com
AIDD kick-off meeting
University of Luxembourg // Luxembourg Slide 2
Luxembourg // 26.01.2021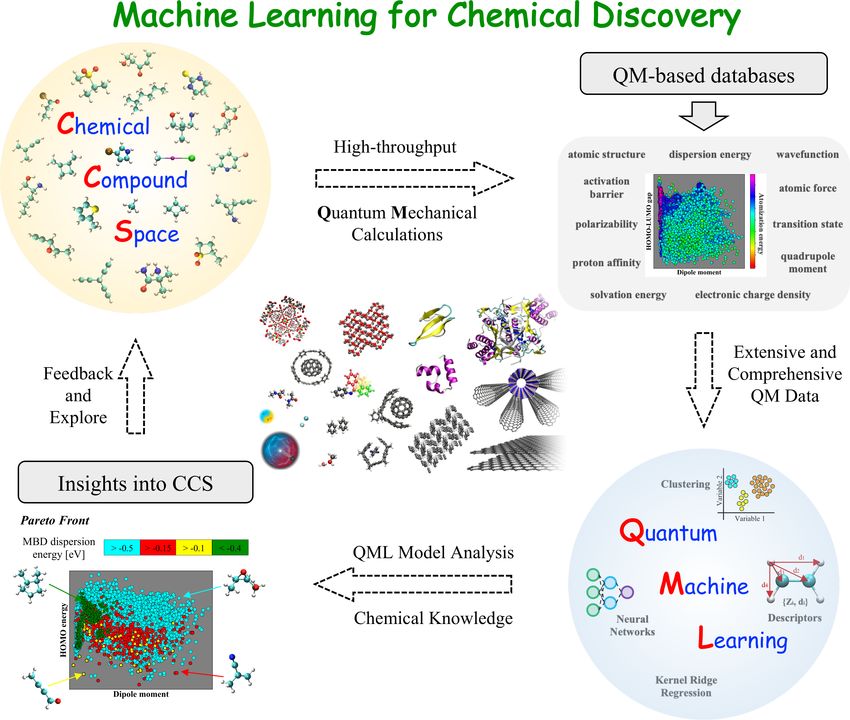
ESR13: Quantum machine learning for reactivity
Molecular property prediction
QM7-X dataset
K.T. Schütt, .., A. Tkatchenko, Nat. Commun., 8, 13890, (2017).
S. Chmiela, .., A. Tkatchenko, Nat. Commun., 9, 3887, (2018).
J. Hoja, .., A. Tkatchenko, Scientific Data, accepted, (2021).
AIDD kick-off meeting
University of Luxembourg // Luxembourg Slide 3
Luxembourg // 26.01.2021
ESR13: Quantum machine learning for reactivity
Dataset generation
Ø Small organic molecules up to 7 heavy atoms (QM7-X dataset)
Ø Small and large molecules with pharmaceutical relevance (UniLu-Janssen dataset)
Ongoing project in collaboration with Janssen Pharmaceutical.
J. Hoja, .., A. Tkatchenko, Scientific Data, accepted, (2021).
AIDD kick-off meeting
University of Luxembourg // Luxembourg Slide 4
Luxembourg // 26.01.2021
0.21 0.15 eV first excitation energy (ZINDO)
0.18 0.13 eV electron affinity (ZINDO/s)
7al/mol on the
0.074 This
0.071is followedauby the analysis of the importance
excitation of the two-
energy at maximal and (ZINDO)
absorption
An angle repre- three-body molecular features and the conclusion.
ESR13: Quantum machine learning for reactivity
1.29 1.26 eV maximal absorption intensity (ZINDO)
ar distributions
0.21 0.18 eV ionization potential (ZINDO/s)
h kernel ridge
d in bold.
Bayesian ridge ■
INVARIANT MANY-BODY INTERACTION
DESCRIPTORS
ules.
redicting QM descriptors
Several Properties Calculated at the B3LYP/6-31G(2df,p) Level of Quantum
chine learning) We represent a molecule or material by the respective finite set
Ridge Regression Trained on 5000 Random Molecules and Tested on the Remaining 126722
ension of BOB
ween
from
Ø Coulomb
pairwise
Journal of Chemical
Journalwhichofthe moleculeTheory
Chemical
matrix
Theory and Computation
or unit and
cell isComputation
constructed. Predicting
Article
Journalatomization
of Chemical Theoryenergy
and Computation
Article
on the GDB-7
F2B Table 4. Predictionunit
F2B + F3B Errors of the B3LYP/6-31G(2df,p) description Table 5. M
2 kcal/mol4.8 on Atomization
1.5 Energy of the Molecules
kcal/mol ofenergy
internal the Setat 0GDB-9
K by 6-31G(2d
es.10 4.8 Various
1.5 ML Models with Random internal
kcal/mol 5K Train Molecules
energy and the
at 298.15 K Large Mo
representation
4.8 Remaining
1.5 126722 Molecules as Test
kcal/mol Setata 298.15 K
enthalpy
Faber et al.
4.8 by 1.5 kcal/mol free energy at 298.15 K train/te
method features MAE RMSE MAX. DEV
atures. For4.7one 3.6 kcal/mol energy of highest occupied molecular orbital small/s
n GDB-7 using
6.0
0.74 kcal/mol
7.9
mean
5.1
RR
6.2
kcal/mol
CM
kcal/mol
185
235
235 occupied1544
energy of lowest
308 between LUMO
gap, difference
molecular orbital
1289 and HOMO PERSPECTIVES small/l
large/s
RR Bohr3 BOB 89 134 653
ns) have 0.72
been 0.49 isotropic polarizability large/l
0.67 RR
0.61 Debye F2B 6.8
dipolemolecule10
moment 462
a
Figure
RR 1. Illustration of the bag-of-bonds
2 F2B + F3B 1.6 similarity.
2.8 extent 88 The The mode
predicting the 7.3 9.0
distance between Bohr
two atoms electronic
of the leftmolecular
molecule spatial
gets isdirectly compared molecules,
0.18an Ralgorithms KNN executed
of ethene: A on
of quantum computers. molecular
the same graphs led 2.4to= 36.9, the publication of 0.4 QM9
Figure 1. Coulomb matrix representation three-dimensional
CM 239 structure
279 converted to a numerical Coulomb matrix using
898
which
atomicreach
coordinates
0.10
to an arbitrary distancekcal/mol the right moleculezero corresponding
point vibrational energy
to
i and nuclear charges Zi. The matrix is dominated by entries resulting from heavy atoms (carbon self-interaction 0.5·6 are marked
g two
samples andin As
carbon 0.40
atoms such,
a distance 0.42
atom
of QML
types
1.33 models
Ø Two- and three-body interactions
KNN
composing
Å result GHz the aim
BOB to provide
pairwise
in ((6.6)/(1.33/0.529))
231 Theamatrixconstant
14.3).rotational
= interaction.
272
contains Adata
one rowsets
758 that
per atom, collectandequilibrium
is symmetric, requires structures
no explicit bond
0.12 information. KNN F2B 151 177 556
pectively. feedback 0.13
KNN
mechanism GHzbetween QM and/or
rotational constant and
B properties of many thousands of Table 6. B
ree-body 0.046 (e.g., SM, and 0.050 GHz F2B + F3B 25 42
rotational constant C 358
(statistical) ML. CMGiven sufficient small molecules (QM7 and QM9) , 47,48 0.15 the Eleme
0.21 KRR
0.12 (Gauss) cal/(mol K) 17
heat capacity 22at 298.15 K 181
es to construct
propose
rked in bold.novel reference data obtained from QM and SM
KRR (Laplace) CM 7.9 10 their129molecular-dynamics trajectories
Figure 3. Mean absolute errors of several electronic ground- and
bond
excited-state properties of the molecules of the set GDB-7 predicted
iant molecular simulations, queries of properly trained
KRR (Gauss) BOB 11 16 (MD17)253 49
and non- withequilibrium molecular
MAE (eV)
KRR using the descriptors CM, BOB, F2B, 0.06and F2BX+ F3B. For CM H−
KRR (Laplace) BOB 4.0 6.0 132
m dis- ML
i with atomic
e pairwise number models
Zi can
KRR (Gauss)
yield accurate
rotational invariant properties
F2B
two-body and three-body
4.8 6.4
structures (ANI-1)
interaction
45
terms 50
andp, . One
BOB, thecan alsokernel
Figure
Laplace calculate
7. has
Mean absolute
been used; forerror of predicting
F2B and the PBE0 atomization
F2B + F3B, the H−
1,23,34
e. Then, a physical system which will
39 define our invariant many-body interaction descriptors. energy of The
the molecules of the set GDB-7 with KRR
of in dependence of
N
In partic- within milliseconds
KRR (Laplace) —F2Bas opposed 8.2to the 11 equilibrium
190 structures and properties
Gauss kernel has been
the
used.
number
ofof training samples. The errors are given
MAEs are normalized by the MAE
in kcal/mol. For
H−
esri}of
i=1.arbitrary
From this physical Invariant Two-Body Interaction Descriptors F . We
the KRR-CM model. 0.025
interaction many CPU hours
KRR (Gauss) orthedays F2B +necessary
F3B 1.5to solve 2.8 solids9651–53, or generate equilibrium
2B
CM and andBOB, the Laplace kernel has been used; for F2B and F2B + F3B, H−
ps 2. Threedescriptors
to alleviate
Figure define
different permutationally invariant set of
representations translational
of andfrom
a molecule derived rotational
its Coulombinvariant two-body
matrix C: (a) eigenspectrum of the C−
Coulomb matrix, (b)the
cordingly, our sortedcorresponding
Coulomb matrix, (c) setquantum
KRR (Laplace) F2B + F3BandCoulomb
interaction terms
of randomly sorted statistical
4.5 matrices. 6.4 non-147equilibrium molecular dynamics the Gauss (MD)kernel has been used. The model hyperparameters have been
C−
ral, Z j , ···is, rnot
j1 , which j , Zmechanics
j ) Figurea
The errors
2. problemsare givenof
Illustration for
inthe representative
kcal/mol.
F2B of and The models used
F3Bsetmolecule are ridge
similarity. data Ffor
Forregression a single element (for example,
2B, the
determined by 10-fold cross-validation.
2BZ2.4 −m C−
The1 main diagonal ≔ ∥ − ∥
k of the Coulomb matrix 0.5
ir ,consists ofrk-nearest
Coulomb matrices, which all follow a slightly different
k
(RR), kernel ridgep (
regression Z , r , Z
(KRR), ) and r neighbors (KNN).
ptors allow for
a polynomial fit ofcompounds. pairwise
the nuclear (1)charges Because
distances of the
to themtotal 1of the
left
energies 2rigorous
1 moleculeof 2 sorting corresponding
1 of atoms.2 Allasilicon)
to fixed
of thempair . The ultimate
54ofexplained in(2)
are goal of QML is to
more detail O−
ny-body
2
inter- while
the free atoms, atom
the types areelements
remaining computed contain intothe a feature entry, which gets compared to
below.
interpolation of QML in complex
+ non- linear 1 develop a universal and efficient model for 0.005 O−
tuple
NoteCoulomb of k repulsion
that atomic numbers
fixed the same
for each 3B
ppair feature
of where
(r1entry
nuclei Z1m
, in ther2∈ n,molecule.
, of the , . r3For
Z 2right , molecule ≔2.2.1.
Z3a) given set
composed of nidentically
Eigenspectrum different atomic
for the numbers
Representation. In the eigenspectrum
100 1,000 10,000 100,000
spaces and their An consequently
≔ {Zi}i=1 with Zicontrolled ∈ {1, ···,13n},the let23Swhole CCS the that
set ofenables the accurate
m1 the eigenvalue
m2 m3problem Cv = λv for each
ionExcept for homometric
term,
asthethey canand be the structures
same
partial
m
pair
1 m2of
,(not, mpresent
3atom in the
types. data set)
Similarly, ≠ZF ∥
3Bj ∀ i, j 12
compares r
representation ∥ ∥ r
three ∥ ∥
bondedr ∥ atoms
2B denote Number of molecules used for training
N−
Coulomb matrix is a which unique have
representation
anallangle. of molecules. Coulomb
40 (Z , Z ) with Z ≤Z and Z , Z ∈ A . Let M denote the
matrix C is solved to represent each molecule as a a
that predictive ·θaccuracy Z3,,≔ the door has i now description 1 ,...,λof
d ), molecules and materials on
of the sequences withouttranslations, tuples λi ≥ λi+1. This
Used for c
essionThe or factdeep rotations, (Z1set , Z 2M , and ∥ri12
symmetry
{1, ∥ j
, ∥
2, r ∥
···,
13 , ∥
vector
r
15}. ∥ j of sorted
)
23 For a given
i j eigenvalues n
physical
(λ2B
(4)
system S, the
2 CM DTNN FCHL NN
set {1,2,···,N} and
opened the for1.an extensive analysis of theand study equal footing and possibly leads to new
operations
size input such
data. as mirror reflections of a molecule
2B in 3D representation (first introduced by Rupp et al. ) is invariant
Table Prediction Errors PBE0 Atomization Energy ofrows BoB SLATM enn-s2s HIP-NN
sy space keep only
1extensible
if and the totalif the energy
two constant two-body
is reflected
∈
interaction
by
+ the
≔
descriptors
with respect
−
F2B
to are now
permutations given
of the by and columns of the
invariance
K.
otherwise.Hansen, of theto
.., of
A.
The number of these
Coulomb where
the
Tkatchenko, interpolated
m
Molecules
matrix1K.-R., m , m
of
with2 Müller, 3the
respect spaces,Set
,
, r ij which
GDB-7
toJ. Chem.
these r i by
Theory was
r for
Various
CoulombjComput., i, j
matrix.=
ML9, insights
{1,2,3},
Models
(2013).
and with on
the CCS underlying regularity and BAML aSLATM MTM SchNet (50% imp
rmative
operations. higher- bond angle indicator
F ≔ { f ( S )} Computing the eigenspectrum of a molecule reduces the HDAD SOAP MBD WaveletsAdditional
W. Pronobis,
setO.A.
ge However,
G(k,von N)there
interactions is are previously
A. Tkatchenko,
N !twoRandom
. problems impossible
K.-R.
5K Train
with the 2B
Müller, dueJ.
Molecules
representation Z , pto
Chem. 2B the
a ̅ m Nat. of m prohibitive
Theory
and Comput.,
Mthe Remaining
∈ dimensionality
2B , Z̅ ∈ S2B
14,
from (3d−6)chemical
1868 degreesrelationships.
(2018).
of freedom(3) to just Reorganizing
d. In the ConstSize
Lilenfeld, K.-R. Müller, A. Tkatchenko, Rev. Chem., 4, (2020).
molecules by their(Ncomputational Molecules
− k) ! matrices, which
Coulomb ⎧cost
as Test
1, make
Set
d of
ESR13: Quantum machine learning for reactivity
Defining novel QM descriptors
Ø Structural properties
• Atomic positions
• Moment of inertia tensor
Ø Molecular properties
• PBE0 atomization energy
• MBD dispersion energy
• HOMO-LUMO gap
• Dipole moment
• Molecular polarizability
• Molecular polarizability tensor
• C6 coefficient
Pr r
op ela
e r t io
Ø Atom-in-a-molecule properties
ty n
Total atomic forces
-p sh
•
ro ip
• Hirshfeld charges
pe
• Hirshfeld dipole moment
rt
y
• Atomic polarizabilities
• vdW radii
AIDD kick-off meeting
University of Luxembourg // Luxembourg Slide 6
Luxembourg // 26.01.2021
ESR13: Quantum machine learning for reactivity
Machine learning for chemical discovery
A. Tkatchenko, Nat. Commun., 11, 4125, (2020).
AIDD kick-off meeting
University of Luxembourg // Luxembourg Slide 7
Luxembourg // 26.01.2021
ESR13: Quantum machine learning for reactivity
Work plan for the doctoral student
Reinforcing knowledge about
chemical reactivity, machine
learning, QM, and SM.
Generation of an extensive QM dataset NEB and/or
using high level of electronic structure. String method
In coordination with
ESR4, ESR5, ESR7.
Development and validation of Secondment at AALTO
machine learning models for predicting (ESR9).
In coordination with TS geometries and energies.
Janssen Pharmaceutical
(month 25-42).
Publishing results in scientific
journals and attending conferences.
ML toolbox for predicting TS geometries and energies.
Getting Doctor degree at UniLu.
AIDD kick-off meeting
University of Luxembourg // Luxembourg Slide 8
Luxembourg // 26.01.2021
Many thanks for the attention. AIDD kick-off meeting University of Luxembourg // Luxembourg Slide 9 Luxembourg // 26.01.2021

You can also read

















































