Theoretical Investigation of Geometries and Bonding of Indium Hydrides in the In2Hx and In3Hy (x = 0-4,6; y = 0-5) Series
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
molecules
Article
Theoretical Investigation of Geometries and Bonding of Indium
Hydrides in the In2Hx and In3Hy (x = 0–4,6; y = 0–5) Series
Anton S. Pozdeev, Pavel Rublev, Steve Scheiner and Alexander I. Boldyrev *
Department of Chemistry and Biochemistry, Utah State University, 0300 Old Main Hill,
Logan, UT 84322-0300, USA
* Correspondence: a.i.boldyrev@usu.edu
Abstract: Boron hydrides have been an object of intensive theoretical and experimental investigation
for many decades due to their unusual and somewhat unique bonding patterns. Despite boron being
a neighboring element to carbon, boron hydrides almost always form non-classical structures with
multi-center bonds. However, we expect indium to form its interesting molecules with non-classical
patterns, though such molecules still need to be extensively studied theoretically. In this work, we
investigated indium hydrides of In2 Hx (x = 0–4,6) and In3 Hy (y = 0–5) series via DFT and ab initio
quantum chemistry methods, performing a global minimum search, chemical bonding analysis, and
studies of their thermodynamical stability. We found that the bonding pattern of indium hydrides
differs from the classical structures composed of 1c-2e lone pairs and 2c-2e bonds and the bonding
pattern of earlier investigated boron hydrides of the Bn Hn+2 series. The studied stoichiometries are
characterized by multi-center bonds, aromaticity, and the tendency for indium to preserve the 1c-2e
lone pair.
Keywords: indium hydrides; bonding; AdNDP analysis; indium compounds
1. Introduction
Citation: Pozdeev, A.S.; Rublev, P.;
Classical organic chemistry is based on a carbon atom in a valence state of IV. This
Scheiner, S.; Boldyrev, A.I. Theoretical
element usually tends to form 2c-2e bonds with carbon and other elements. Despite boron
Investigation of Geometries and
and carbon being neighboring elements, boron tends to form multi-center bonds [1–3]. In
Bonding of Indium Hydrides in the
particular, it was shown [4] that the Bn Hn+2 classical structures become less stable along
In2 Hx and In3 Hy (x = 0–4,6; y = 0–5)
Series. Molecules 2023, 28, 183.
the series because boron avoids expected sp2 -hybridization. Despite the rich chemistry of
https://doi.org/10.3390/
boron hydrides being well-studied [5–9], our knowledge of indium hydride compounds
molecules28010183
is minimal. Only a small number of indium hydrides and their derivatives have been
previously synthesized [10,11] or theoretically investigated [12–14]. To our knowledge,
Academic Editor: Demeter Tzeli
almost no conformational search procedures have been applied for indium hydrides with
Received: 23 November 2022 several indium atoms; therefore, the possible non-trivial properties of indium hydrides
Revised: 21 December 2022 have been missed.
Accepted: 23 December 2022 Being a relatively heavy element, indium atoms have less preference for any type of
Published: 26 December 2022 sp hybridization in hydrogen compounds compared to boron and aluminum. One of the
main reasons for this is related to weaker hydrogen–indium orbital overlaps and, therefore,
lower interaction: dissociation energies are 3.42 eV and 2.48 eV for B-H and In-H bonds,
respectively [15]. Another important reason is a more significant energy gap between 5s and
Copyright: © 2022 by the authors.
5p orbitals induced by complete electronic 4d-subshell and the subsequent nucleus charge
Licensee MDPI, Basel, Switzerland.
screening effect. It can be vividly seen from the difference between excitation energies 2 Po
This article is an open access article
→ 4 P of boron and indium atoms [16,17].
distributed under the terms and
Thus, are the indium and boron hydride structures similar or different? Are classical
conditions of the Creative Commons
Attribution (CC BY) license (https://
arrangements with only 1c-2e lone pairs and 2c-2e bonds possible for indium hydrides? In
creativecommons.org/licenses/by/
this work, we investigated the nature of In2 Hx (x = 0–4,6) and In3 Hy (y = 0–5) compounds
4.0/).
using the Coalescence Kick global optimization techniques, chemical bonding AdNDP
Molecules 2023, 28, 183. https://doi.org/10.3390/molecules28010183 https://www.mdpi.com/journal/molecules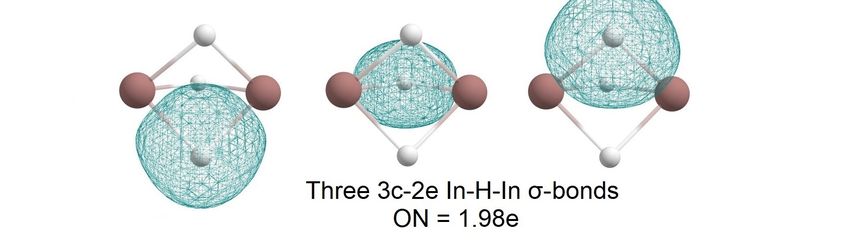
Thus, are the indium and boron hydride structures similar or different? Are classical
arrangements with only 1c-2e lone pairs and 2c-2e bonds possible for indium hydrides?
Molecules 2023, 28, 183 2 of 12
In this work, we investigated the nature of In2Hx (x = 0–4,6) and In3Hy (y = 0–5) compounds
using the Coalescence Kick global optimization techniques, chemical bonding AdNDP
analysis, thermodynamic stability toward H2 dissociation to answer the abovementioned
questions.
analysis, thermodynamic stability toward H2 dissociation to answer the abovementioned
questions.
2. Results and Discussions
2. Results and Discussions
2.1. Global Geometry Optimization and Bonding Analysis
2.1. Global Geometry Optimization and Bonding Analysis
Initially, we performed the CK search for In2Hx and In3Hy (x = 0–4; y = 0–5). The in-
Initially, we performed the CK search for In2 Hx and In3 Hy (x = 0–4; y = 0–5). The
vestigated global minima structures are shown in Figure 1. The obtained geometry of the
investigated global minima structures are shown in Figure 1. The obtained geometry of
global minimum and energy ordering for low-lying isomers of In2H2 stoichiometry is in
the global minimum and energy ordering for low-lying isomers of In2 H2 stoichiometry is
entire agreement with the previous investigation [18]. Other low-lying geometries are
in entire agreement with the previous investigation [18]. Other low-lying geometries are
given in the SI file (Figures S1–S11).
given in the SI file (Figures S1–S11).
Figure 1.
Figure Global minimum
1. Global minimum structures
structures of
of (a)
(a) In
In22H
Hxx(x(x==0–4),
0–4),(b)
(b)InIn3H
3H (y(y= =
yy 0–5).
0–5).
According to
According to the
the AdNDP
AdNDP analysis,
analysis, completely
completely unsaturated
unsaturated structures
structures In and In
In22 and In33
have similar bonding patterns. In the case of the In 2 molecule in the first triplet
have similar bonding patterns. In the case of the In2 molecule in the first triplet state, we state, we
found two 1c-2e lone pairs, one 2c-1e σ-bond with ON = 0.99e, and one 2c-1e
found two 1c-2e lone pairs, one 2c-1e σ-bond with ON = 0.99e, and one 2c-1e π-bond with π-bond with
ON = 0.99e, where “ON” stands for occupancy number, and “e” reflects that occupancy
ON = 0.99e, where “ON” stands for occupancy number, and “e” reflects that occupancy
number is related to the number of electrons.
number is related to the number of electrons.
For In , we investigated three 1c-2e lone pairs with ON = 1.80e, 3c-2e π-bond with
For In33, we investigated three 1c-2e lone pairs with ON = 1.80e, 3c-2e π-bond with
ON = 2.00e, and 3c-1e σ-bond with ON = 0.98e for an unpaired electron. We assign a
ON = 2.00e, and 3c-1e σ-bond with ON = 0.98e for an unpaired electron. We assign a mol-
molecule as being doubly aromatic (i.e., π and σ aromatic; Figure 2). Negative NICSZZ
ecule as being doubly aromatic (i.e., π and σ aromatic; Figure 2). Negative NICSZZ values
values at different distances from XY-plane can be considered another argument for the
at different distances from XY-plane can be considered another argument for the aroma-
aromaticity of In3 [19]. For example, in the case of benzene NICSZZ (0) = −15.199 ppm,
ticity of In3 [19]. For example, in the case of benzene NICSZZ(0) = −15.199 ppm, NICSZZ(1)
NICSZZ (1) = −29.517 ppm, NICSZZ (2) = −17.315 ppm, NICSZZ (3) = −8.014 ppm
= −29.517 ppm, NICSZZ (2) = −17.315 ppm, NICSZZ (3) = −8.014 ppm showing the presence
showing the presence of π-aromaticity. In the case of In3 NICSZZ (0) = −2.146 ppm,
of π-aromaticity. In the case of In3 NICSZZ(0) = −2.146 ppm, NICSZZ(1) = −17.565 ppm,
NICSZZ (1) = −17.565 ppm, NICSZZ (2) = −17.139 ppm, NICSZZ (3) = −9.760 ppm. For
NICSZZ(2) = −17.139 ppm, NICSZZ(3) = −9.760 ppm. For the In3 cluster, the absolute values
the In3 cluster, the absolute values of NICSZZ (0) and NICSZZ (1) are much smaller than
of NICS
those ZZ(0) and NICSZZ(1) are much smaller than those for benzene. However, the values
for benzene. However, the values of NICSZZ (2) and NICSZZ (3) are similar for both
of NICS ZZ(2) and NICSZZ(3) are similar for both molecules; an explanation of the difference
molecules; an explanation of the difference may be based on the types of involved orbitals.
Benzene is a π aromatic molecule, but In3 is a doubly π and σ aromatic, which indicates the
significant difference of NICSZZ (0) and NICSZZ (1) values near the molecular plane, where
σ orbitals influence more significantly. It is worth mentioning that an unpaired electron
on a σ-like orbital provides a more energetically stable state than a state with an unpaired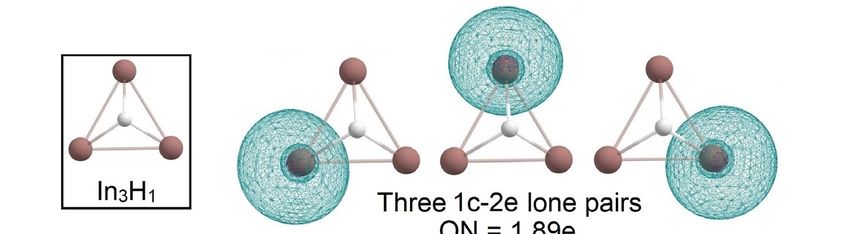
may be
may be based
based on
on the
the types
typesofof involved
involved orbitals.
orbitals. Benzene
Benzene isisaaππ aromatic
aromatic molecule,
molecule,
is a doubly π and σ aromatic, which indicates the significant
is a doubly π and σ aromatic, which indicates the significant difference of difference of NICS
NICSzzzz(
Molecules 2023, 28, 183 NICS zz(1) values near the molecular plane, where σ orbitals influence
NICSzz(1) values near the molecular plane, where σ orbitals influence more signifi 3 more
of 12 signif
It is worth mentioning that an unpaired electron on a σ-like orbital provides aamor
It is worth mentioning that an unpaired electron on a σ-like orbital provides mor
getically stable
getically stable state
statethan
thanaa state
state with
withan an unpaired
unpaired electron
electron on on aaπ-like
π-like orbital.
orbital. The
The
ence
electron on a π-like between
ence orbital.
between those
Thethose electronic
difference between
electronic states
those
states is 4.12 kcal/mol
electronic
is 4.12 kcal/molstates and
and was
is 4.12 obtained
waskcal/mol
obtained usingusing
and was obtained CCSD(T)/cc-pVTZ(-PP)
CCSD(T)/cc-pVTZ(-PP) level of
using the ∆-CCSD(T)/cc-pVTZ(-PP)
level of theory.
theory. Theofsame
level
The same
theory. procedure
The same
procedure was performed for
procedure
was performed for
molecule.
was performedmolecule.
for the InAnAn alternative electronic state with two occupied perpendicular singl
2 molecule.
alternative Anelectronic
alternative electronic
state with two state with two
occupied occupied single
perpendicular
pied 2c-1e
2c-1e π-bonds
perpendicular single-occupied
pied π-bonds2c-1einπ-bonds
in the Slater
the Slater determinant
indeterminant was investigated.
the Slater determinant
was investigated. This state
state was
was investigated.
This was less
less
by 5.8
This state was less
by 5.8 kcal/mol
stable by 5.8at
kcal/mol at theΔ-CCSD(T)/cc-pVTZ(-PP)
kcal/mol
the Δ-CCSD(T)/cc-pVTZ(-PP)
at the ∆-CCSD(T)/cc-pVTZ(-PP) level of
level of theory.
theory.
level of theory.
Figure 2.
Figure 2. The chemical
Figure 2. The chemical
bonding
The chemical bonding
patternbonding pattern
of In2 and of In
In3 global
pattern of In22minimum
and In
and In33 global
global minimum
structures structures
obtained
minimum by
structures obta
obtai
AdNDPanalysis.
AdNDP analysis.AdNDP analysis.
Further “hydrogenation”
Further evolution of the In
Further “hydrogenation”
“hydrogenation” 2 Hx series
evolution
evolution ofreveals
of the In2some
the In features
2Hx series
Hx series of indium
reveals
reveals some features
some feature
hydride compounds.
dium Besides
hydride 1c-2e lone pairs,
compounds. in In2 H
Besides 1c-2e
1, weloneobserved
pairs, 3c-2e
in In 2In-H-In
H σ-bond 3c-2e I
1, we observed
dium hydride compounds. Besides 1c-2e lone pairs, in In2H1, we observed 3c-2e I
with occupationσ-bond
numberwith ON occupation
= 2.00e andnumber
2c-1e In-In 2.00ewith
ONσ-bond ON
2c-1e=In-In
0.98e,σ-bond
but in In 2 H2ON
σ-bond with occupation number ON == 2.00e and 2c-1e
and In-In with
σ-bond with ON == 0.9
0.9
we did not find 2c-2e
in In
In22Hbonds—only
H22 we
we did
did not two
not find 1c-2e lone
find 2c-2e pairs
2c-2e bonds—onlyand
bonds—onlytwo two 3c-2e
two 1c-2e In-H-In
1c-2e lone σ-bonds
lone pairs
pairsand
and twowith
two3c-2e
3c-2e In-H
In-H
in
ON = 2.00e (Figure
bonds 3).with
Thus,
ONupon
= 2.00e“hydrogenation,”
(Figure 3). Thus, indium
upon saves its lone pair
“hydrogenation,” instead,
indium saves its lo
bonds with ON = 2.00e (Figure 3). Thus, upon “hydrogenation,” indium saves its lon
forming the 2e-2c classical
instead, In-In σ-bond.
forming the 2e-2c classical In-In σ-bond.
instead, forming the 2e-2c classical In-In σ-bond.
Figure 3. The chemical bonding pattern of In2 H1 and In2 H2 global minimum structures obtained by
Figure3.
3.The
Thechemical
chemicalbonding
bondingpattern
patternof
ofIn
In22H
H11and
andIn
In22H
H22global
globalminimum
minimumstructures
structuresobta
obta
AdNDP analysis.Figure
AdNDP analysis.
AdNDP analysis.
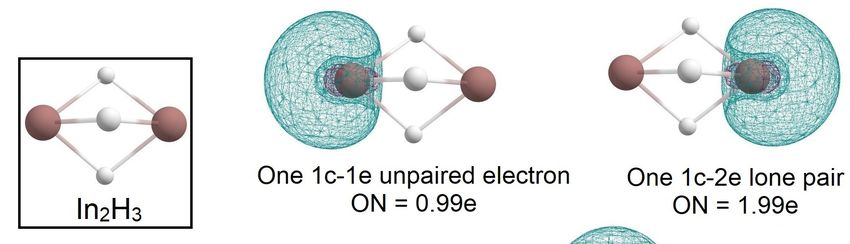
In the case of the In2 H3 molecule, the AdNDP algorithm allowed us to describe the
In the
the case
case of
of the
the In
In22H
H33 molecule,
molecule, the
the AdNDP algorithm
algorithm allowed
allowed us us to descr
bonding pattern as In three 3c-2e In-H-In σ-bonds with ONAdNDP
= 1.98 e, one 1c-1e unpaired to descri
bonding
bonding pattern
pattern as three
asindium 3c-2e
three 3c-2e In-H-In
In-H-In σ-bonds with ON = 1.98 e, one 1c-1e unpaire
electron with ON = 0.99e on one atom, and aσ-bonds with
1c-2e lone ON
pair on= another
1.98 e, one 1c-1e unpaire
indium
tron with
with ON ON = 0.99e
0.99e on
on one
one indium
indium atom,
atom, and
and aa 1c-2e
1c-2e lone
lone pair
pair on
on another
another indium
indium
atom with ON tron
= 1.99e (Figure=4).
with ON = 1.99e (Figure 4).
Accordingwith
to [4],ON = 1.99ea(Figure
structure in Figure4). 5 is the global minimum of B2 H4 . In the present
study, structure b (Figure 5) is the global minimum for In2 H4 . Among others, one significant
difference between these two molecules is that in the case of B2 H4, there is one B-B 2c-2e
σ-bond and no 1c-2e lone pairs, but In2 H4 has two 3c-2e In-H-In σ-bonds with ON = 1.98e,
one 1c-2e lone pair on one indium atom, and no In-In 2c-2e σ-bonds (Figure 6). Moreover,
our attempts to find a similar In2 H4 structure to the structure in Figure 5a at different
theoretical levels were unsuccessful. Thus, this direct comparison of the global minimum
structures of B2 H4 and In2 H4 demonstrates the difference in indium and boron hydride
bonding patterns. Both have non-classical structures (i.e., they have delocalized multi-
center 2e bonds). However, all boron valence electrons participate in bonding, whereas
indium tends to save its lone pair.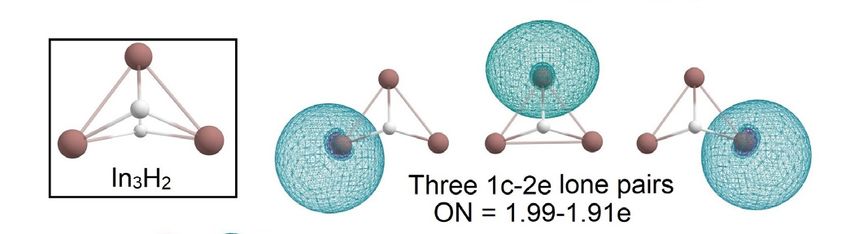
Molecules
Molecules2022,
2023,27,
28,x183
FOR PEER REVIEW 4 4ofof12
12
Figure 4. The chemical bonding pattern of the In2H3 global minimum structure obtained by AdNDP
analysis.
According to [4], structure a in Figure 5 is the global minimum of B2H4. In the present
study, structure b (Figure 5) is the global minimum for In2H4. Among others, one signifi-
cant difference between these two molecules is that in the case of B2H4, there is one B-B 2c-
2e σ-bond and no 1c-2e lone pairs, but In2H4 has two 3c-2e In-H-In σ-bonds with ON =
1.98e, one 1c-2e lone pair on one indium atom, and no In-In 2c-2e σ-bonds (Figure 6).
Moreover, our attempts to find a similar In2H4 structure to the structure in Figure 5A at
different theoretical levels were unsuccessful. Thus, this direct comparison of the global
minimum structures of B2H4 and In2H4 demonstrates the difference in indium and boron
hydride bonding patterns. Both have non-classical structures (i.e., they have delocalized
multi-center
Figure4.4.The
Figure
2e bonds).
Thechemical
chemical However,
bonding
bonding patternall
pattern of
boron
ofthe
the In22HH3valence
In
electrons
globalminimum
3global minimum participate
structure
structure
in by
obtained
obtained
bonding,
byAdNDP
AdNDP
whereas
analysis.
analysis. indium tends to save its lone pair.
According to [4], structure a in Figure 5 is the global minimum of B2H4. In the present
study, structure b (Figure 5) is the global minimum for In2H4. Among others, one signifi-
cant difference between these two molecules is that in the case of B2H4, there is one B-B 2c-
2e σ-bond and no 1c-2e lone pairs, but In2H4 has two 3c-2e In-H-In σ-bonds with ON =
1.98e, one 1c-2e lone pair on one indium atom, and no In-In 2c-2e σ-bonds (Figure 6).
Moreover, our attempts to find a similar In2H4 structure to the structure in Figure 5A at
different theoretical levels were unsuccessful. Thus, this direct comparison of the global
minimum structures of B2H4 and In2H4 demonstrates the difference in indium and5boron
Molecules 2022, 27, x FOR PEER REVIEW of(b)
12
Figure
Figure5.5.The
Thecomparison
comparisonofofthe global
the globalminimum structures for
for(a)
(a)BB
2HH4 (according to [4]) and
hydride bonding patterns. Both haveminimum structures
non-classical structures (i.e., (according
2 4 they have to [4]) and
delocalized (b)
InIn
2H4.
H .
2 4
multi-center 2e bonds). However, all boron valence electrons participate in bonding,
whereas indiumtheoretical
An earlier tends to save
studyits of
lone pair. and non-classical structures of B2H4 in [4]
classical
showed that the non-classical structure is more stable by 2.9 kcal/mol. In this work, the
CK search allowed us to find a classical structure for In2H4. The AdNDP analysis reveals
that both indium atoms are connected via 2c-2e σ-bonds with ON = 2.00e, and all other In-
H bonds are 2c-2e σ-bonds with ON = 1.99e. The energy difference between the classical
and non-classical structures is 10.8 kcal/mol at the QRO-CCSD(T)/cc-pVTZ(-PP)//U-
TPSSh/def2-TZVPP level of theory. The energy difference is too big to suggest a competi-
tion of the classical structure with the global minimum structure. The bonding analysis of
the two structures is shown in Figure 6.
Figure 5. The comparison of the global minimum structures for (a) B2H4 (according to [4]) and (b)
In2H4.
An earlier theoretical study of classical and non-classical structures of B2H4 in [4]
showed that the non-classical structure is more stable by 2.9 kcal/mol. In this work, the
CK search allowed us to find a classical structure for In2H4. The AdNDP analysis reveals
that both indium atoms are connected via 2c-2e σ-bonds with ON = 2.00e, and all other In-
H bonds are 2c-2e σ-bonds with ON = 1.99e. The energy difference between the classical
and non-classical structures is 10.8 kcal/mol at the QRO-CCSD(T)/cc-pVTZ(-PP)//U-
TPSSh/def2-TZVPP level of theory. The energy difference is too big to suggest a competi-
tion of the classical structure with the global minimum structure. The bonding analysis of
the two structures is shown in Figure 6.
Figure 6. The chemical bonding pattern of In H4 —non-classical structure (global minimum) and
Figure 6. The chemical bonding pattern of In22H4―non-classical structure (global minimum) and
classicalstructure
classical structureobtained
obtainedby
byAdNDP
AdNDPanalysis.
analysis.
Additionally, we decided to investigate the In2H6 species as an analog of the famous
diborane molecule. The CK algorithm found only one non-dissociated structure of this
stoichiometry; it totally resembles the structure of B2H6. It indicates the stability of the
motif. The structure and bonding analysis are presented in Figure 7. We found four 2c-2e
In-H σ-bonds with ON = 2.00e and two 3c-2e In-H-In σ-bonds with ON = 1.97e.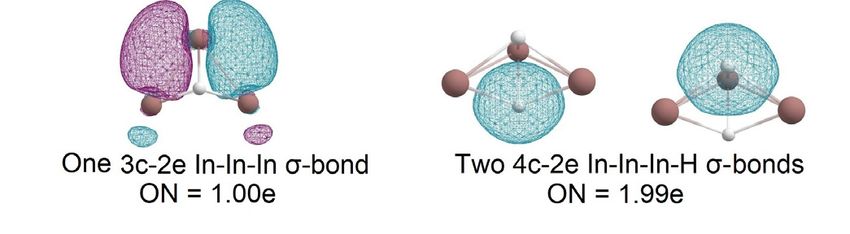
Molecules 2023, 28, 183 5 of 12
An earlier theoretical study of classical and non-classical structures of B2 H4 in [4]
showed that the non-classical structure is more stable by 2.9 kcal/mol. In this work,
the CK search allowed us to find a classical structure for In2 H4 . The AdNDP analysis
reveals that both indium atoms are connected via 2c-2e σ-bonds with ON = 2.00e, and
all other In-H bonds are 2c-2e σ-bonds with ON = 1.99e. The energy difference between
the classical and non-classical structures is 10.8 kcal/mol at the QRO-CCSD(T)/cc-pVTZ(-
PP)//U-TPSSh/def2-TZVPP
Figure level ofoftheory.
6. The chemical bonding pattern The energy difference
In2H4―non-classical is toominimum)
structure (global big to suggest
and
a competition
classical structureofobtained
the classical structure
by AdNDP with the global minimum structure. The bonding
analysis.
analysis of the two structures is shown in Figure 6.
Additionally,
Additionally,we wedecided
decidedto toinvestigate
investigatethetheIn
In22HH6 6species
speciesasasan ananalog
analogofofthe
thefamous
famous
diborane
diboranemolecule.
molecule.The TheCK CKalgorithm
algorithmfound
foundonly
onlyoneonenon-dissociated
non-dissociatedstructure
structureofofthis
this
stoichiometry;
stoichiometry;itittotally
totallyresembles
resemblesthe thestructure
structureof of BB22HH6.6 .ItItindicates
indicatesthe
thestability
stabilityofofthe
the
motif.The
motif. Thestructure
structureand
andbonding
bondinganalysis
analysisarearepresented
presentedininFigure Figure7.7.We
Wefound
foundfour
four2c-2e
2c-2e
In-Hσ-bonds
In-H σ-bondswithwithON ON==2.00e
2.00eand
andtwo
two3c-2e
3c-2eIn-H-In
In-H-Inσ-bonds
σ-bondswith withON
ON==1.97e.
1.97e.
Thechemical
Figure7.7.The
Figure chemicalbonding
bondingpattern
patternofofthe
theInIn2H
2H 6 global
6 global minimumstructure
minimum structureobtained
obtainedby
byAdNDP
AdNDP
analysis.
analysis.
InInthe
theInIn
3H 3 yHseries,
y series,
wewe observed
observed the gradual
the gradual “modification”
“modification” of theofInthe In3 triangular
3 triangular clus-
cluster upon “hydrogenation”.
ter upon “hydrogenation.” In3H and3 In3H2 haveIn H and In H have similar structures where
3 2similar structures where each hydrogen each hy-
isdrogen
connected is connected
with the In with the In3 cluster
3 cluster via
via 4c-2e In-In-In-H
4c-2e In-In-In-H σ -bond with σ -bond
ON = with1.99e ON = 1.99e
(Figure 8).
(Figure 8). In 3 H retains σ-aromaticity with an occupation number close
In3H retains σ-aromaticity with an occupation number close to ideal 2.00e. Calculated val- to ideal 2.00e. Cal-
culated
ues of NICSvalues of NICS areforNICS
ZZ for In3H1ZZ
In3 HZZ1(0)
are=NICS
+3.804ZZ (0)
ppm,= +3.804
NICSZZ ppm,
(1) =NICS ZZ (1)
−12.155 = −12.155
ppm, NICSZZppm,
(2)
NICS ZZ (2) = − 14.271 ppm, NICS ZZ (3) = − 8.235 ppm. A positive
= −14.271 ppm, NICSZZ(3) = −8.235 ppm. A positive value near the center of a molecule value near the center of a
molecule
with a rapidwith a rapidofdecrease
decrease of NICS
NICSzz value as zz value
the as theincreases
distance distancecanincreases can beof
be evidence evidence
σ-aro-
of σ-aromaticity [20]. Due to hydrogen atom incorporation, the absolute
maticity [20]. Due to hydrogen atom incorporation, the absolute values of NICSzz of In3H1 values of NICSzz
of In3 H1 are smaller than for In3 . For In3 H2 , we found an interesting bonding pattern of
are smaller than for In3. For In3H2, we found an interesting bonding pattern of 3c-1e In-In-
3c-1e In-In-In σ-bond with ON = 1.00e; the AdNDP analysis reveals that it consists of two
px orbitals and one perpendicular py orbital.
The 4c-2e In-In-In-H σ-bond appeared to be stable for the In3 Hy series. In the In3 H3
cluster, the bonding motif of the In3 H2 structure was saved, and the third H atom was
introduced in the cluster as a bridged atom in a 3c-2e In-H-In σ-bond with ON = 1.99e.
This bonding pattern allowed In3 H3 to keep three 1c-2e lone pairs on each indium atom
with ON = 1.98–1.85e. Further “hydrogenation” leads to In3 H4 losing one 4c-2e In-In-In-H
σ-bond and one 1c-2e lone pair. It has two 1c-2e lone pairs, one 2c-1e In-In σ-bond with
ON = 0.99e, one 2c-1e In-H σ-bond with ON = 0.99e, two 3c-2e In-H-In σ-bonds with
ON = 1.99e, and one 4c-2e In-In-In-H σ-bond with ON = 1.97e (Figure 9).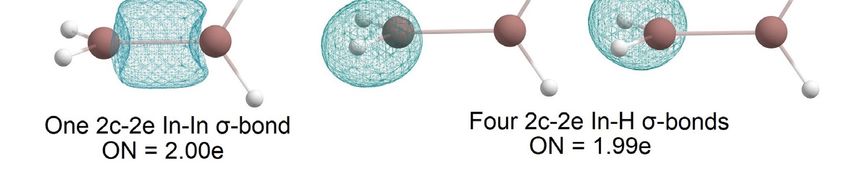
Molecules 2022, 27, x FOR PEER REVIEW 6 of 12
Molecules 2023, 28, 183 6 of 12
In σ-bond with ON = 1.00e; the AdNDP analysis reveals that it consists of two px orbitals
and one perpendicular py orbital.
Molecules 2022, 27, x FOR PEER REVIEW 7 of 12
Figure 8. The chemical bonding pattern of In3 H1 and In3 H2 obtained by AdNDP analysis.
Figure 8. The chemical bonding pattern of In3H1 and In3H2 obtained by AdNDP analysis.
The 4c-2e In-In-In-H σ-bond appeared to be stable for the In3Hy series. In the In3H3
cluster, the bonding motif of the In3H2 structure was saved, and the third H atom was
introduced in the cluster as a bridged atom in a 3c-2e In-H-In σ-bond with ON = 1.99e.
This bonding pattern allowed In3H3 to keep three 1c-2e lone pairs on each indium atom
with ON = 1.98‒1.85e. Further “hydrogenation” leads to In3H4 losing one 4c-2e In-In-In-H
σ-bond and one 1c-2e lone pair. It has two 1c-2e lone pairs, one 2c-1e In-In σ-bond with
ON = 0.99e, one 2c-1e In-H σ-bond with ON = 0.99e, two 3c-2e In-H-In σ-bonds with ON
= 1.99e, and one 4c-2e In-In-In-H σ-bond with ON = 1.97e (Figure 9).
The
Figure9.9.The
Figure chemical
chemical bonding
bonding pattern
pattern ofofInIn
3H33Hand
3 and
In3In
H34H4 obtained
obtained by by AdNDP
AdNDP analysis.
analysis.
As in the case of In2 H4 and B2 H4 , the global minimum structures of In3 H5 and B3 H5 [4]
As in the case of In2H 4 and B2H4, the global minimum structures of In3H5 and B3H5 [4]
are different. Both systems are presented in Figure 10. B3 H5 has three 2c-2e B-H terminal
are different. Both systems are presented in Figure 10. B3H5 has three 2c-2e B-H terminal
σ-bonds, two bridged B-H-B σ-bonds, and three B atoms that are bonded via 3c-2e B-B-B σ-
σ-bonds, two bridged B-H-B σ-bonds, and three B atoms that are bonded via 3c-2e B-B-B
bond and a 3c-2e B-B-B π-bond, so there are no 1c-2e lone pairs again. In In H , two indium
σ-bond and a 3c-2e B-B-B π-bond, so there are no 1c-2e lone pairs again.3 In5 In3H5, two
indium atoms save 1c-2e lone pairs; there is one In-H 2c-2e terminal σ- bond with ON =
1.99e, three bridged 3c-2e In-H-In σ-bonds with ON = 1.98–1.97e, and one 4c-2e In-In-In-
H σ-bond with ON = 1.96e (Figure 11). Thus, for boron clusters, aromaticity is found for
both saturated and non-saturated structures [21], but for indium, we observed aromaticity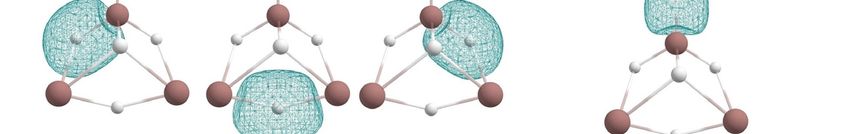
Figure 9. The chemical bonding pattern of In3H3 and In3H4 obtained by AdNDP analysis.
As in the case of In2H4 and B2H4, the global minimum structures of In3H5 and B3H5 [4]
Molecules 2023, 28, 183 7 of 12
are different. Both systems are presented in Figure 10. B3H5 has three 2c-2e B-H terminal
σ-bonds, two bridged B-H-B σ-bonds, and three B atoms that are bonded via 3c-2e B-B-B
σ-bond and a 3c-2e B-B-B π-bond, so there are no 1c-2e lone pairs again. In In3H5, two
atomsatoms
indium save 1c-2e
savelone pairs;
1c-2e lonethere
pairs;is there
one In-H 2c-2e
is one terminal
In-H σ- bond with
2c-2e terminal ON with
σ- bond = 1.99e,
ONthree
=
bridged
1.99e, three3c-2e In-H-In
bridged σ-bonds
3c-2e In-H-Inwith ON = with
σ-bonds 1.98–1.97e,
ON = and one 4c-2e
1.98–1.97e, In-In-In-H
and one 4c-2eσ-bond with
In-In-In-
H ON = 1.96e
σ-bond with(Figure
ON =11). Thus,
1.96e for boron
(Figure clusters,
11). Thus, for aromaticity is found
boron clusters, for bothissaturated
aromaticity found for and
non-saturated structures [21], but for indium, we observed aromaticity only
both saturated and non-saturated structures [21], but for indium, we observed aromaticity for the most
unsaturated
only for the mosthydrides. It is another
unsaturated hydrides.demonstration
It is another ofdemonstration
the difference between the patterns
of the difference be- of
boron and indium hydrides.
tween the patterns of boron and indium hydrides.
Molecules 2022, 27, x FOR PEER REVIEW 8 of 12
BnHn+2 series [4] (i.e., classical structures become progressively less stable along the series).
The bonding pattern of the classical structure of In3H5 is similar to what was observed for
the classical
Figure
Figure TheThe
10.10. structure ofofInthe
comparison
comparison Hthe
2of ; it global
hasminimum
4global two types
minimum ofstructures
bonds—2c-2e
structures 3H5In-H
forB(a)
for (a) 3 H5 σ-bond
B(according with
(according ON
toand
to [4]) [4]) =
and
(b)
1.99e
In H5.Inand
3(b) 3 H5 .2c-2e In-In σ bond with ON = 1.99e.
The low-lying classical In3H5 structure (Figure 11) is about 17.2 kcal/mol higher than
the global minimum structure; this corresponds to the trend previously observed for
Figure11.
Figure Thechemical
11.The chemicalbonding
bondingpattern
patternofofInIn3H
3H 5 —non-classical
5―non-classical
structure(global
structure (globalminimum)
minimum)and
and
classicalstructure.
classical structure.
The low-lying
2.2. Thermodynamic classical
Stability In3 H5 structure (Figure 11) is about 17.2 kcal/mol higher
Observation
than the global minimum structure; this corresponds to the trend previously observed for
Bn HTo estimate the thermodynamics stability of the indium hydrides toward H2 dissoci-
n+2 series [4] (i.e., classical structures become progressively less stable along the series).
ation, we calculated the potential energy difference of locally reoptimized structures at
QRO-CCSD(T)/cc-pVTZ(-PP)//U-TPSSh/def2-TZVPP level of theory. The results of these
calculations are shown in Table 1.
According to the data, almost all indium hydrides are stable toward H2 dissociation,
except In2H3, In2H6, and In3H4. However, even in the case of these molecules, the ΔE of the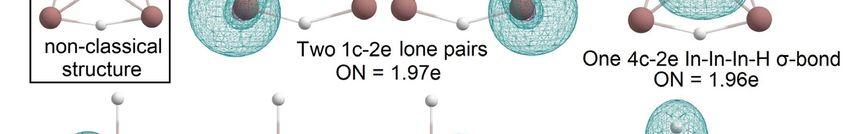
Molecules 2023, 28, 183 8 of 12
The bonding pattern of the classical structure of In3 H5 is similar to what was observed
for the classical structure of In2 H4 ; it has two types of bonds—2c-2e In-H σ-bond with
ON = 1.99e and 2c-2e In-In σ bond with ON = 1.99e.
2.2. Thermodynamic Stability Observation
To estimate the thermodynamics stability of the indium hydrides toward H2 disso-
ciation, we calculated the potential energy difference of locally reoptimized structures at
QRO-CCSD(T)/cc-pVTZ(-PP)//U-TPSSh/def2-TZVPP level of theory. The results of these
calculations are shown in Table 1.
Table 1. Analysis of thermodynamic stability toward H2 dissociation at the QRO-CCSD(T)/cc-pVTZ(-
PP)//U-TPSSh/def2-TZVPP level of theory.
Stoichiometry Decomposition Product Stability Eproducts − Ereagents , kcal/mol
In2 H2 In2 + H2 stable 25.2
In2 H3 In2 H + H2 unstable −0.1
stable 4.8
In2 H4 In2 H2 + H2 In2 + 2H2
stable 30.0
unstable −2.1
In2 H4 + H2 In2 H2 +
In2 H6 stable 2.7
2H2 In2 + 3H2
stable 27.9
In3 H2 In3 + H2 stable 12.1
In3 H3 In3 H + H2 stable 12.2
unstable −0.5
In3 H4 In3 H2 + H2 In3 + 2H2
stable 11.6
According to the data, almost all indium hydrides are stable toward H2 dissociation,
except In2 H3 , In2 H6 , and In3 H4 . However, even in the case of these molecules, the ∆E of
the dissociation reaction is very small.
3. Theoretical Methods
The global minimum search was carried out using the Coalescence Kick (CK) algorithm
written by Averkiev [22,23] to find a global minimum and corresponding low-lying isomers
for In2 Hx and In3 Hy (x = 0–4,6; y = 0–5). The basic workflow of the CK algorithm consists of
(1) the random placing of atoms in a sizeable Cartesian box, (2) the shift of atoms toward the
center of mass until they coalesce up to the pairwise sums of pre-defined covalent radii of
atoms, (3) the standard local geometry optimization using Ab Initio, DFT, or semi-empirical
approach. The CK procedure is repeated several thousand times to obtain an adequate
statistic, and the most stable structure is implied to be a global minimum. The CK original
code is available online at the open repository [24].
All calculations utilizing the CK procedure were carried out using unrestricted Kohn–
Sham formalism with density functional MN-15. It was chosen for its appropriate applica-
bility to various electronic structure problems, including multireference behavior [25]. The
LANL2DZ basis set was chosen for its balance of speed and accuracy [26]; additionally, the
Hay–Wadt pseudopotential was applied to indium atoms to account for scalar relativity
correction [27]. The specific generation size of random structures for the CK method for
each stoichiometry is shown in the SI file (Table S1). Gaussian 16 was used as the main
driver for the local optimization step [28] in the CK algorithm.
After the CK global minimum search, all obtained low-lying isomers in the 15 kcal/mol
energy window from the global minimum were reoptimized with a more accurate def2-
TZVPP basis set [29] and TPSSh density functional [30], which tends to provide better
energy ordering of isomers but cannot be generously applied to any electronic problem.
Obtained local minima were verified by nuclear Hessian calculation with the same method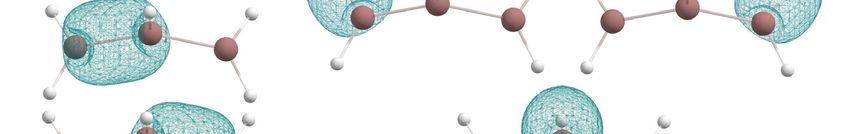
Molecules 2023, 28, 183 9 of 12
and basis set. The re-optimization step and all subsequent single-point calculations utilized
the ORCA 5.03 suite [31,32].
Using obtained local minima geometries, single point energies were recalculated at
the CCSD(T) level of theory with the cc-pVTZ [33] basis set on hydrogen atoms and the
cc-pVTZ-PP basis set on indium atoms complemented with the SK-MCDHF-RSC pseu-
dopotential [34,35]. The CCSD(T) approach is a gold standard for the accurate prediction
of energy ordering and provides meaningful results only in conjunction with large basis
sets. Quasi-restricted orbitals formalism (QRO-CCSD(T)) was employed to eliminate the
influence of spin-contamination in the open-shell coupled clusters calculations. In addition,
all CCSD(T) reference UHF wavefunctions were tested using UHF/UHF stability analysis
based on the CIS method [36].
Thus, the final energy ordering of low-lying isomers was estimated using QRO-
CCSD(T)/cc-pVTZ energies and U-TPSSh/def2-TZVPP geometries and corresponding
zero-point energy corrections (ZPE). This calculation scheme will be denoted as “QRO-
CCSD(T)/cc-pVTZ(-PP)//U-TPSSh/def2-TZVPP”.
It should be outlined that, due to the adiabatic treatment of the potential energy
surface, the CK algorithm can be applied only for one spin state at a time. For stoichiometry
with an even number of electrons, we chose a singlet multiplicity in the algorithm; for an
odd number of electrons, we decided to use a doublet multiplicity. To verify that other spin
states are not global minima, they were locally reoptimized in the corresponding triplet
or quartet multiplicities at the U-TPSSh/def2-TZVPP level of theory. Further single point
energy calculations at the QRO-CCSD(T)/cc-pVTZ(-PP) theoretical level demonstrated that
in the case of all stoichiometries, except In2 and In3 H1 , the energy difference between a
global minimum structure in the lowest spin multiplicity and a corresponding low-lying
structure in triplet (or quartet) multiplicity was at least 15–30 kcal/mol. That allowed us to
justify the choice of multiplicity in the CK algorithm.
In the case of In2, we found the triplet state to be more stable than the singlet state by
6.8 kcal/mol, which is in accordance with previous findings [37]. For In3 H1 , the singlet
state is more stable than the locally reoptimized triplet state; however, the energy difference
between them is relatively small (~2.5 kcal/mol). Therefore, we carried out an additional
CK search to find the actual global minimum of the triplet state. In this way, we found
the singlet state still to be more stable than the triplet state by 1.4 kcal/mol at the QRO-
CCSD(T)/cc-pVTZ(-PP) level of theory. The energy differences between spin states are
provided in the SI file (Table S2).
Chemical bonding for all global minimum isomers for each stoichiometry was ana-
lyzed using the adaptive natural density partitioning (AdNDP) algorithm implemented in
AdNDP 2.0 [38,39] as an effective method of deciphering bonding in molecular clusters
with untrivial electron delocalization. This approach is based on the Lewis idea that an
electron pair is the main bonding element. The algorithm leads to the partitioning of the
electron density into elements with the lowest symmetry-appropriate number of atomic
centers per electron pair, which allows for the representation of an electronic structure as
n-center two-electron bonds (nc-2e, n is an interval from one to the total number of atoms
participating in the bond). The same procedure may be applied to open-shell systems and
may recover nc-1e bonding elements. Despite the usage of the “electron pair idea,” AdNDP
and its ideological predecessor NBO analyses based on the density matrix representation of
the wavefunction; therefore, near doubly occupied bonding elements are obtained through
chains of similarity or unitary transformations of canonical KS “wavefunction,” which
is represented by single Slater determinant. Thus, the limit of two-electron occupancy
per bonding element is not arbitrary and is dictated by Pauli’s exclusion principle for
fermions and the primary modern approach to compose many-body wavefunctions. Thus,
the AdNDP algorithm recovers classical Lewis bonding elements (i.e., 1c-2e lone-pairs and
2c-2e bonds) and the delocalized bonding elements similar to occupied canonical molecular
orbitals, which are the most appropriate for this study. In this work, the density matrices
Molecules 2023, 28, 183 10 of 12
for the AdNDP were obtained at the U-TPSSh/def2-TZVPP level of theory. Visualization
of molecular structures and AdNDP orbitals was performed with ChemCraft [40].
In addition, the AdNDP algorithm may show molecules’ potential aromatic or anti-
aromatic character. To verify it, the components of nucleus-independent chemical shift
(NICSzz) corresponding to the principal z-axis perpendicular to a ring plane may be used
as a good characteristic of aromaticity [40]. We calculated NICSzz(R) values (R means the
distance from the center of a ring in Å units) at the TPSSh/def2-TZVPP level of theory.
4. Conclusions
In this work, we used the CK algorithm for the global minimum search for In2 Hx and
In3 Hy (x = 0–4,6; y = 0–5) stoichiometry. Found global minimum geometries were used to
investigate the structural evolution of In2 and In3 clusters under “hydrogenation”. We also
investigated their chemical bonding pattern using the AdNDP algorithm and tested their
thermodynamic stability toward H2 dissociation. Our analysis revealed that both boron
and indium hydrates have non-classical structures with multi-center 2e bonds, and they
are much more stable than classical structures with sp2 hybridization. Indium hydrides are
characterized by non-intuitive global minimum geometries, where indium tends to save
its lone pair even if it leads to the loss of a classical 2c-2e bond. Some unsaturated indium
clusters have aromatic properties. We want to emphasize that the revealed difference
between boron and indium hydrides is closely related to the difference in the fundamental
properties of boron and indium atoms: higher excitation energy of indium atoms and less
energetically favorable indium–hydrogen bond formation.
We hope this work will inspire further theoretical and experimental investigation of
these exotic indium species.
Supplementary Materials: The following supporting information can be downloaded at: https:
//www.mdpi.com/article/10.3390/molecules28010183/s1, Table S1. Population size used for CK
algorithm for In2 Hx and In3 Hy (x = 0–4,6; y = 0–5) stoichiometries; Table S2. Spin-state energy
difference; Table S3. In2 Hx and In3 Hy (x = 0–4,6; y = 0–5) global minimum geometries for singlet
and doublet states found the lowest lying isomers for triplet and quartet states (XYZ coordinates);
Table S4. NICSzz values; Figures S1–S11. Global minimum geometries and low-lying geometries
with corresponding relative energies for each In2 Hx and In3 Hy (x = 0–4,6; y = 0–5) stoichiometry,
chemical bonding patterns for global minimum geometries.
Author Contributions: Conceptualization: A.S.P., S.S., P.R. and A.I.B.; methodology: A.S.P., S.S.,
P.R. and A.I.B.; writing—original draft preparation: A.S.P., S.S., P.R. and A.I.B.; writing—review,
and editing: S.S. and A.I.B.; visualization: A.S.P. All authors have read and agreed to the published
version of the manuscript.
Funding: The research was funded by the R. Gaurth Hansen Professorship fund to A.I.B.
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Data Availability Statement: The data that support the findings of this study are available from the
corresponding authors upon reasonable request.
Acknowledgments: The support and resources from the Centre for High Performance Computing at
the University of Utah are gratefully acknowledged.
Conflicts of Interest: The authors declare no conflict of interest.
Sample Availability: Samples of the compounds are not available from the authors.
References
1. Lipscomb, W.N. Boron Hydrides; Benjamin Inc.: New York, NY, USA, 1963.
2. Muetterties, E.L. Boron Hydride Chemistry; Academic Press: New York, NY, USA, 1975.
3. Pan, S.; Barroso, J.; Jalife, S.; Heine, T.; Asmis, K.R.; Merino, G. Fluxional Boron Clusters: From Theory to Reality. Acc. Chem. Res.
2019, 52, 2732–2744. [CrossRef] [PubMed]Molecules 2023, 28, 183 11 of 12
4. Osorio, R.; Olson, J.K.; Tiznado, W.; Boldyrev, A.I. Analysis of Why Boron Avoids sp2 Hybridization and Classical Structures in
the Bn Hn+2 Series. Eur. J. Chem. 2012, 18, 9677–9681. [CrossRef] [PubMed]
5. Cho, B.T. Recent development and improvement for Boron hydride-based catalytic asymmetric reduction of unsymmetrical
ketones. Chem. Soc. Rev. 2009, 38, 443–452. [CrossRef] [PubMed]
6. Sivaev, I.B.; Bregadze, V.V. Polyhedral Boranes for Medical Applications: Current Status and prospects. Eur. J. Inorg. Chem. 2009,
2009, 1433–1450. [CrossRef]
7. Fakioğlu, E.; Yürüm, Y.; Veziroğlu, T.N. A review of hydrogen storage systems based on Boron and its compounds. Int. J.
Hydrogen Energy 2004, 29, 1371–1376. [CrossRef]
8. Sergeeva, A.P.; Popov, I.A.; Piazza, Z.A.; Li, W.L.; Romanescu, C.; Wang, L.S.; Boldyrev, A.I. Understanding Boron through
Size-Selected Clusters: Structure, Chemical Bonding, and Fluxionality. Acc. Chem. Res. 2014, 47, 1349–1358. [CrossRef]
9. Jian, T.; Chen, X.; Li, S.D.; Boldyrev, A.I.; Li, J.; Wang, L.S. Probing the structures and bonding of size-selected Boron and
doped-Boron clusters. Chem. Soc. Rev. 2019, 48, 3550–3591. [CrossRef]
10. Wiberg, E.; Schmidt, M.Z. Uber Wasserstoff-Verbindungen des Indiums. Z. Nat. B 1957, 12, 54. [CrossRef]
11. Miyai, T.; Inoue, K.; Yasuda, M.; Shibata, I.; Baba, A. Preparation of a novel Indium hydride and application to practical organic
synthesis. Tetrahedron Lett. 1998, 39, 1929–1932. [CrossRef]
12. Hunt, P.; Schwerdtfeger, P. Are the Compounds InH3 and TlH3 Stable Gas Phase or Solid State Species? Inorg. Chem. 1996, 35,
2085–2088. [CrossRef]
13. Schwerdtfeger, P.; Heath, G.A.; Dolg, M.; Bennett, M.A. Low Valencies and Periodic Trends in Heavy Element Chemistry. A
Theoretical Study of Relativistic Effects and Electron Correlation Effects in Group 13 and Period 6 Hydrides and Halides. Am.
Chem. Soc. 1992, 114, 7518–7527. [CrossRef]
14. Liu, Y.; Duan, D.; Tian, F.; Liu, H.; Wang, C.; Huang, X.; Li, D.; Ma, Y.; Liu, B.; Cui, T. Pressure-Induced Structures and Properties
in Indium Hydrides. Inorg. Chem. 2015, 54, 9924–9928. [CrossRef] [PubMed]
15. Huber, K.P.; Herzberg, G. Chapter IV Constants of Diatomic Molecules. In Molecular Spectra and Molecular Structure, 1st ed.;
Springer: New York, NY, USA, 1979.
16. Edlén, B.; Ölme, A.; Herzberg, G.; Johns, J.W.C. Ionization Potential of Boron, and the Isotopic and Fine Structure of 2s2 p2.2 D. J.
Opt. Soc. Am. 1970, 60, 889. [CrossRef]
17. Paschen, F.; Campbell, J.S. Das erste Funkenspektrum des Indiums in II. Ann. Phys. 1938, 423, 29–75. [CrossRef]
18. Mulla, H.F.; Franke, P.R.; Sargenta, C.; Douberlya, G.E.; Turney, J.M.; Schaefer, H.F., III. Four isomers of In2 H2 : A careful
comparison between theory and experiment. Mol. Phys. 2021, 119, e1979675. [CrossRef]
19. Báez-Grezab, R.; Ruizc, L.; Pino-Rios, R.; Tiznado, W. Which NICS method is most consistent with ring current analysis?
Assessment in simple monocycles. RSC Adv. 2018, 8, 13446–13453. [CrossRef]
20. Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.V.R. Nucleus-Independent Chemical Shifts (NICS) as an
Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [CrossRef]
21. Sergeeva, A.P.; Averkiev, B.B.; Zhai, H.-J.; Boldyrev, A.I.; Wang, L.S. All-Boron analogues of aromatic hydrocarbons: B17 − and
B18 −. J. Chem. Phys. 2011, 134, 224304. [CrossRef]
22. Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.J.; Wang, L.S. All-Boron aromatic clusters as potential new inorganic ligands and
building blocks in chemistry. Coord. Chem. Rev. 2006, 250, 2811–2866. [CrossRef]
23. Saunders, M. Stochastic search for isomers on a quantum mechanical surface. J. Comput. Chem. 2004, 25, 621–626. [CrossRef]
24. Coalescence Modification to Saunders KICK Version. Available online: https://kick.science/KICK_Saunders_coalescence.html
(accessed on 25 November 2022).
25. Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad
accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [CrossRef]
[PubMed]
26. Dunning, T.H.; Hay, P.J. Modern Theoretical Chemistry; Schaefer, H.F., III, Ed.; Plenum: New York, NY, USA, 1977; Volume 3, pp.
1–28.
27. Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J.
Chem. Phys. 1985, 82, 284. [CrossRef]
28. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.;
Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
29. Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn:
Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [CrossRef] [PubMed]
30. Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized
Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [CrossRef]
31. Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [CrossRef]
32. Neese, F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 12, e1606.
[CrossRef]
33. Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms Boron through neon and hydrogen. J.
Chem. Phys. 1989, 90, 1007. [CrossRef]Molecules 2023, 28, 183 12 of 12
34. Metz, B.; Stoll, H. Small-core multiconfiguration-Dirac–Hartree–Fock-adjusted pseudopotentials for post-d main group elements:
Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563. [CrossRef]
35. Peterson, K.A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the
post-d group 13–15 elements. J. Chem. Phys. 2003, 119, 11099. [CrossRef]
36. Bauernschmitt, R.; Ahlrichs, R. Stability analysis for solutions of the closed shell Kohn–Sham equation. J. Chem. Phys. 1996,
104, 9047. [CrossRef]
37. Balasubramanian, K.; Li, J. Spectroscopic properties and potential energy surfaces of In2 . J. Chem. Phys. 1998, 88, 4979. [CrossRef]
38. Tkachenko, N.V.; Boldyrev, A.I. Chemical bonding analysis of excited states using the Adaptive Natural Density Partitioning
method. Phys. Chem. Chem. Phys. 2019, 21, 9590–9596. [CrossRef] [PubMed]
39. Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem.
Chem. Phys. 2008, 10, 5207–5217. [CrossRef] [PubMed]
40. Chemcraft. Available online: http://www.chemcraftprog.com (accessed on 25 November 2022).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual
author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to
people or property resulting from any ideas, methods, instructions or products referred to in the content.You can also read

















































