The proliferative and apoptotic activities of E2F1 in the mouse retina
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Oncogene (2001) 20, 7073 ± 7084
ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/01 $15.00
www.nature.com/onc
The proliferative and apoptotic activities of E2F1 in the mouse retina
Suh-Chin J Lin1, Stephen X Skapek2, David S Papermaster3, Mark Hankin4 and Eva Y-HP Lee*,1
1
Department of Molecular Medicine/Institute of Biotechnology, University of Texas Health Science Center at San Antonio, Texas,
TX 78245-3207, USA; 2Department of Hematology/Oncology, St. Jude Children's Research Hospital, Memphis, Tennessee,
TN 38105, USA; 3Department of Neuroscience, University of Connecticut Health Science Center, Farmington, Connecticut,
CT 06030-3401, USA; 4Department of Anatomy and Neurobiology, Medical College of Ohio, Toledo, Ohio, OH 43614, USA
The E2F1 transcription factor controls cell proliferation (Adams and Kaelin, 1995; Slansky and Farnham,
and apoptosis. E2F1 activity is negatively regulated by 1996). Genetic studies in both Drosophila (Duronio et
the retinoblastoma (RB) protein. To study how inactiva- al., 1995) and mammalian cells (Ishizaki et al., 1996;
tion of Rb and dysregulated E2F1 aects the developing Wu et al., 1996) demonstrate that E2F activity is
retina, we analysed wild-type and Rb7/7 embryonic required for cell proliferation. In addition, overexpres-
retinas and retinal transplants and we established sion of E2F1 has been shown to be sucient to drive
transgenic mice expressing human E2F1 in retinal quiescent cells into S phase in vitro (Johnson et al.,
photoreceptor cells under the regulation of the IRBP 1993). E2F1 cooperates with activated Ras to trans-
promoter (TgIRBPE2F1). A marked increase in cell form ®broblasts (Johnson et al., 1994; Singh et al.,
proliferation and apoptosis was observed in the retinas of 1994; Xu et al., 1995). In mice, expression of E2F1 in
Rb7/7 mice and TgIRBPE2F1 transgenic mice. In the the presence of activated Ras results in skin tumor
transgenic mice, photoreceptor cells formed rosette-like formation (Pierce et al., 1998a). Interestingly, in
arrangements at postnatal days 9 through 28. Complete addition to its role in cell proliferation, overexpression
loss of photoreceptors followed in the TgIRBPE2F1 mice of E2F1 but not other E2F proteins also triggers
but not in the Rb7/7 retinal transplants. Both RB- apoptosis in cells. In the absence of p53, E2F1-induced
de®cient and E2F1-overexpressing photoreceptor cells apoptosis is signi®cantly reduced (DeGregori et al.,
expressed rhodopsin, a marker of terminal dierentia- 1997; Kowalik et al., 1995; Qin et al., 1994; Shan and
tion. Loss of p53 partially reduced the apoptosis and Lee, 1994; Wu and Levine, 1994). This apoptosis-
resulted in transient hyperplasia of multiple cell types in promoting property of E2F1 likely contributes to the
the TgIRBPE2F1 retinas at postnatal day 6. Our phenotype of E2F1 de®cient mice, which are defective
®ndings support the concept that cross-talk occurs in thymocyte apoptosis and predisposed to tumors at
between dierent retinal cell types and that multiple an advanced age (Field et al., 1996; Yamasaki et al.,
genetic pathways must become dysregulated for the full 1996).
oncogenic transformation of neuronal retinal cells. An understanding of how E2F activity may be
Oncogene (2001) 20, 7073 ± 7084. regulated in mammalian cells ®rst came from the
®ndings of a functional interaction between E2F1 and
Keywords: E2F1; p53; RB and retinoblastoma the protein product of the retinoblastoma susceptibility
gene Rb (reviewed in Dyson, 1998). RB may block the
transcriptional activity of E2F1 by direct binding to its
Introduction transactivation domain and preventing its interaction
with other components of the transcriptional machin-
The E2F transcription factor was initially identi®ed as ery (Ross et al., 1999). In addition, the presence of
a cellular DNA-binding activity required for the histone deacetylases in the E2F/RB complexes could
adenovirus E1A-mediated activation of the viral E2 result in transcription repression by a mechanism
gene (reviewed in Dyson, 1998). E2F functions as a involving chromatin regulation (Brehm and Kouzar-
heterodimer consisting of an E2F family member, ides, 1999). Finally, other RB-interacting proteins have
E2F1 through 6, and a DP protein, DP1 and DP2 been identi®ed that could mediate transcriptional
(reviewed in Dyson, 1998). E2F can bind to speci®c repression of E2F-dependent genes (Skapek et al.,
regulatory elements in genes required for DNA 2000). The physical interaction between RB and E2F is
synthesis including DNA polymerase a, thymidine detected during the G1 phase of the cell cycle. Upon
kinase and dihydrofolate reductase; and cell cycle phosphorylation of RB by cyclin-dependent kinases,
regulators including cyclin A, cyclin E and cdc2 RB, E2F, and other proteins found in this complex
(like HDAC-1) dissociate to allow the activation of the
E2F responsive genes (reviewed in Dyson, 1998; Zhang
*Correspondence: EY-HP Lee; E-mail: Leee@uthscsa.edu
et al., 1999).
Received 11 August 2000; revised 23 August 2001; accepted 23 Two Rb-related genes p107 and p130 have also been
August 2001 characterized (reviewed in Mulligan and Jacks, 1998).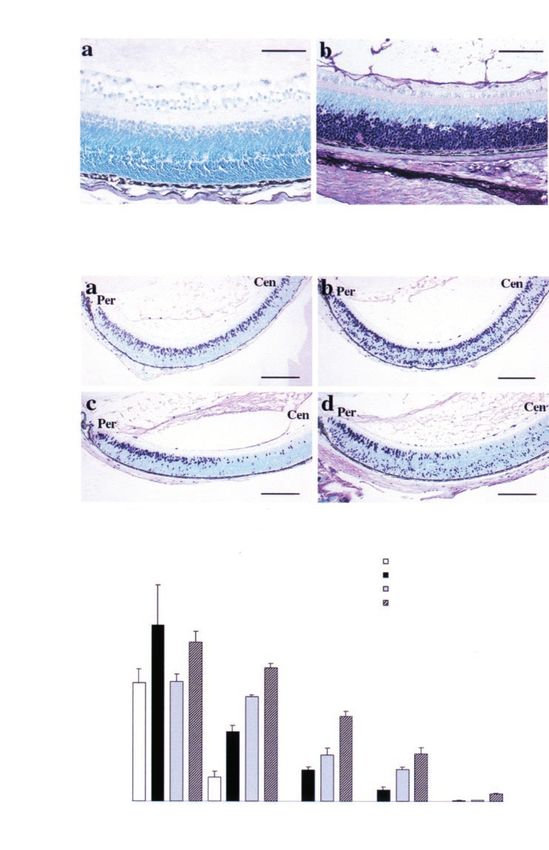
E2F1 and p53 inactivation in retinoblastoma
S-CJ Lin et al
7074
Protein products of both genes interact with E2F and regulation of E2F1 and loss of the p53 tumor
can act as negative regulators of cell proliferation. suppressor contribute, individually or in combination,
Unlike Rb, however, mutations of p107 and p130 are to photoreceptor development and tumor formation.
rarely found in human tumors and mutation of either
p107 or p130 in mice leads to dierent phenotypes
(reviewed in Lin et al., 1996; Mulligan and Jacks,
Results
1998). The dierent biological functions of dierent
RB family members may be due to their association
Proliferation and apoptosis in Rb-deficient retinas
with speci®c E2Fs (reviewed in Dyson, 1998; Nevins,
1998). RB interacts with E2F1, E2F2 and E2F3 during There is aberrant proliferation and extensive apoptosis
late G1 to S phase. In contrast, p107 preferentially in the developing nervous system of RB-de®cient
interacts with E2F4 in proliferating cells during embryos (Lee et al., 1994; Macleod et al., 1996).
S phase whereas p130 binds to E2F4 and E2F5 How RB de®ciency aects retinal development is not
primarily in G0 phase. Despite these distinctions, known, in part due to embryonic lethality of Rb7/7
recent studies of mice lacking more than one member mice around E12.5 ± E14.5 (Lee et al., 1992). To begin
of the Rb gene family have provided evidence that Rb, to study the eects of loss of RB in the retina, we
p107 and p130 have overlapping as well as distinct analysed retinal cell proliferation and apoptosis in
function (see below). retinas from E13.5 wild-type and Rb7/7 embryos. In
That the loss of Rb causes retinoblastoma in humans normal embryos, the inner layers of the retinal
has been well established. On the other hand, how loss ventricular cells did not incorporate BrdU at E13.5
of Rb aects the retina in mouse models is more (Figure 1Aa). In contrast, in Rb7/7 embryos, many
confusing. Homozygous Rb7/7 mouse embryos show cells in the same region continued to enter the cell cycle
ectopic proliferation and excess apoptosis of the (Figure 1Ab). In wild-type embryos, little TUNEL
nervous system and die around embryonic day 14.5 staining was observed in the normal E13.5 retina
(E14.5) (Clarke et al., 1992; Jacks et al., 1992; Lee et (Figure 1Ac), but TUNEL-stained cells were readily
al., 1992). Heterozygous Rb+/7 mice develop pituitary detected in Rb7/7 retina (Figure 1Ad). Examination of
and thyroid tumors (Hu et al., 1994; Jacks et al., 1992; cell proliferation and apoptosis at dierent embryonic
Nikitin and Lee, 1996) and all tumors harbor stages (E11.5 ± E15.5) demonstrated that ectopic BrdU-
mutations of the wild-type Rb allele (Hu et al., 1994; positive cells appeared in the retina before TUNEL-
Nikitin and Lee, 1996); however, retinoblastoma does stained cells (data not shown), a ®nding that is similar
not occur. In contrast, multiple dysplastic lesions to ®ndings in the spinal cord and dorsal root ganglia of
develop in the photoreceptor cell layer of the retina Rb7/7 embryos (Lee et al., 1994). These data indicate
of Rb+/7; p1077/7 mice but not in Rb+/7 or p1077/7 that RB loss causes excessive ectopic cell proliferation
mice (Lee et al., 1996). Retinoblastomas of amacrine and apoptosis in embryonic retinal progenitor layers of
cell origin were observed in chimeric mice harboring the mouse retina.
Rb7/7; p1077/7 cells (Robanus-Maandag et al., 1998). In addition to regulating cell proliferation and
Thus, it seems that dysfunction of both RB and p107 apoptosis, RB is known to facilitate the dierentiation
are required for retinal tumor formation in mice. of certain types of cells (reviewed in Lipinski and
One interpretation of these collective data is that loss Jacks, 1999). Therefore, we sought to determine how
of RB and RB-related proteins releases certain E2Fs loss of RB aected the dierentiation of murine
from suppression to enhance cell proliferation and photoreceptors. In wild-type mice, retina development
tumor formation. Consistent with this, inactivation of continues after birth. Because loss of RB causes
E2F1 reduces tumorigenesis in Rb+/7 mice and extends embryonic lethality, to fully address the eect of RB-
the survival of Rb+/7 mice and Rb7/7 embryos (Tsai de®ciency on retinal development, we analysed retinal
et al., 1998; Yamasaki et al., 1998). Furthermore, E2F1 tissue that had been transplanted into normal neonatal
de®ciency rescues cell death in Rb7/7 embryos in mice. It has previously been shown that transplanted
tissues undergoing p53-dependent apoptosis (Macleod embryonic retinal tissue continues to develop and form
et al., 1996; Morgenbesser et al., 1994) including the laminar structures (Hankin et al., 1993). Following
ocular lens and the central nervous system (Tsai et al., transplantation of retinas from E11.5 ± E12.5 embryos
1998). Taken together, these data provide biochemical into wild-type or Rb+/7 neonates, the transplanted
and genetic support for the regulatory role of RB on retinal tissues formed either sheet-like structures or
E2F1 activities. rosette structures characterized by inside-out, con-
We undertook the present series of experiments to centric foldings of the donor tissue (Figure 1B). The
study the eects of dysregulated E2F1 activity in multi-layered structures were evident in both wild-type
photoreceptors. We accomplished this by studying RB- and Rb-de®cient retinal transplants (Figure 1Ba,b). At
de®cient photoreceptors in Rb7/7 mouse embryos and the cellular level, both wild-type and Rb7/7 retino-
following transplantation of Rb7/7 retinal cells into blasts matured and dierentiated into photoreceptors
normal recipient mice. In addition, we have established as evidenced by expression of opsin (Figure 1Be,f).
a transgenic mouse model in which E2F1 is constitu- Interestingly, using this model of retina development,
tively expressed in maturing photoreceptors. We have we con®rmed that RB loss causes excess apoptosis.
used these studies to further understand how loss of Whereas only a few apoptotic cells were seen in wild-
OncogeneE2F1 and p53 inactivation in retinoblastoma
S-CJ Lin et al
7075
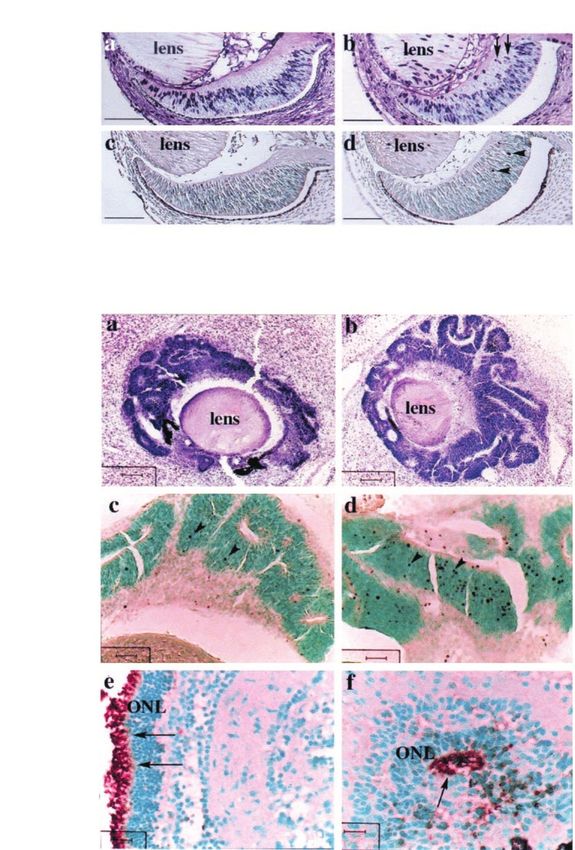
Figure 1 Eects of RB de®ciency on proliferation, apoptosis and dierentiation of retinal cells. (A) Enhanced cell proliferation and
cell death in the retina of Rb7/7 embryos. Pregnant mice were injected with BrdU. BrdU uptake in the retinas of E13.5 embryos
was compared (a and b). Apoptotic cells were shown using TUNEL assay (c and d). Arrows indicate ectopic BrdU-positive cells and
arrowheads indicate apoptotic cells. Scale bar, 100 mm. (B) Cell death and dierentiation in the photoreceptor cells of retinal
transplants. Retinal tissues of E11.5 ± 12.5 embryos were transplanted intracranially into neonatal Rb+/7 or wild-type mice. Sections
of retinal transplants were stained with H&E (a and b), analysed for apoptosis using the TUNEL assay (c and d), or immunostained
with anti-opsin antibodies (e and f). Arrowheads indicate apoptotic nuclei; arrows indicate opsin expression in the inner/outer
segments of the photoreceptor cells. ONL, outer nuclear layer. Scale bar, (a,b) 100 mm, (c,d) 40 mm, (e,f) 15.6 mm. Left panel, wild-
type embryos; right panel, Rb7/7 embryos
type transplants, abundant apoptotic cells were found these results demonstrate that RB may be required for
in Rb7/7 transplants (Figure 1Bc,d). Taken together, ecient cell cycle arrest and prevention of apoptosis in
OncogeneE2F1 and p53 inactivation in retinoblastoma
S-CJ Lin et al
7076
cells in the developing mouse retina, but is not required dierentiating photoreceptors that normally would
for the expression of opsin in photoreceptors. have excited the cell cycle.
Generation of TgIRBPE2F1 transgenic mice Apoptosis of the neuroretinal cells in TgIRBPE2F1
transgenic mice
Because the loss of RB appears to alter retinal
development in these mouse transplant experiments, Despite the increased proliferation observed in retinas
and RB is known to regulate E2F1, we established a from TgIRBPE2F1 mice, there were fewer photorecep-
transgenic mouse model to study the eect of enhanced tor cells in the retinas in adult transgenic mice (Figure
expression of E2F1 in retinas in vivo. To do this, we 5f, discussed below). The decreased number of
generated transgenic mice expressing human E2F1 photoreceptor cells in the retina of the transgenic mice
(hE2F1) cDNA under the regulation of IRBP suggests that overexpression of E2F1 may also result in
promoter (TgIRBPE2F1 mice). This promoter directs cell death. To better characterize the mechanism by
the expression of the transgene in retinal photoreceptor which the photoreceptors were lost, the degree of
cells and pinealocytes (Liou et al., 1990). Several apoptosis was determined during the early stages of
founder mice were identi®ed using PCR and Southern retinal development. In retinas from wild-type mice,
analyses (data not shown) and two hE2F1 transgenic apoptotic cells were only occasionally observed during
lines (IRBP-E37 and -E45) were established. the major period of retinal dierentiation (Figure
To con®rm that the transgene led to ectopic 3A,B; Young, 1984). By contrast, abundant apoptotic
expression of E2F1, we performed immunohistochem- cells were found throughout the ventricular layer at all
ical analyses. In normal mice, E2F1 was detectable in stages in the transgenic mice with greatest apoptosis at
the retina of E18.5 embryos (data not shown), but was P4 and P6 (Figure 3A,B). At later stages, fewer
not detectable after birth (Figure 2A). In the retina of apoptotic cells were seen perhaps in part due to the
transgenic mice, hE2F1 was expressed in the outer previous loss of cells in the ONL in the transgenic
ventricular cell layer, the developing rods, from P0 retinas (Figure 3A). The location of most of the
(day of birth) through P6 (Figure 2A). The hE2F1 was apoptotic cells in the ONL indicates that the dying cells
also present in mature photoreceptor cells at later are, in fact, developing photoreceptors.
stages (data not shown). All ospring from both lines During normal development, the arrest of cellular
had similar expression patterns and developed the same proliferation and the initiation of dierentiation
phenotype (see below). proceeds from the central to the peripheral retina
(Young, 1985). Therefore, we sought to correlate the
time course of proliferation and apoptosis with this
Developing neuroretinal cells in TgIRBPE2F1 transgenic
developmental feature. Proliferation in the central
mice continue to synthesize DNA
retina was most robust at P0, gradually decreased
During retinal development, the ventricular cells cease after birth and ceased at P9 in transgenic mice (Figure
proliferation at P6 in the central region and at P11 in 2C). Interestingly, there were more apoptotic cells in
the peripheral region of the retina (Young, 1985). The the central region compared to the peripheral region in
development of post-mitotic cells parallels morpholo- transgenic retinas earlier in postnatal development
gical changes in the developing outer nuclear layer (Figure 3B), whereas at P9 and P10 there were more
(ONL) of photoreceptors. Active proliferation, as apoptotic cells in the periphery (Figure 3A,B). Hence,
evidenced by BrdU incorporation, was seen throughout it appears that the excess cell proliferation caused by
the retina of the wild-type mice from P0 to P4 (Figure E2F1 precedes excess apoptosis and that the apoptosis
2B,C). During this period, there were more BrdU- appears to occur in a developmentally regulated
positive cells in the retina of transgenic mice than in pattern.
the control retina (Figure 2B,C). By P6, BrdU stained
cells were restricted to the peripheral retina in wild-
Delayed differentiation in the neuroretinal cells in
type mice, whereas BrdU-positive cells were detected in
TgIRBPE2F1 transgenic mice
the central as well as the peripheral retina in the
transgenic mice at P6 (Figure 2B). These data During retinal development, multiple cell types are
demonstrate that forced expression of E2F1 in vivo is generated and arranged into morphologically distinct
sucient to drive DNA synthesis in the developing layers. To assess the eect of E2F1 expression on
retina that should have been ®lled with post-mitotic development of the multi-layer structure of retina,
photoreceptors. Interestingly, the ectopic BrdU-positive retina sections of P4 ± P6 were compared (Figure 4). At
cells in the transgenic mice were dierent from BrdU- P6 the outer plexiform layer (OPL), which is composed
positive cells in the normal mice in two regards. First, of synaptic processes between photoreceptor cells in the
they were located near the pigment epithelium (PE) in ONL and cells in the inner nuclear layer (INL), was
transgenic mice (Figure 2Bb,d) as opposed to the inner evident in the control mice (Figure 4a), but was not
plexiform layer (IPL) in wild-type mice (Figure 2Ba,c). apparent in transgenic mice (Figure 4b). However, the
Second, their nuclei were more rounded than the OPL was visible at P8 and later stages in transgenic
BrdU-positive cells in wild-type mice (data not shown). retinas (Figure 3Af,h) when the cells of the ONL were
Their location and nuclear shape suggest that they are better dierentiated. This ®nding suggests that ectopic
OncogeneE2F1 and p53 inactivation in retinoblastoma
S-CJ Lin et al
7077
Figure 2 Ectopic E2F1 expression enhances retinal cell proliferation. (a) E2F1 expression in the retinas. E2F1 immunostaining was
performed on retinal sections of wild-type (left) and TgIRBPE2F1 retinas (right) of 6-day-old (P6) neonates using anti-E2F1
antibodies. Scale bar, 100 mm. (B) BrdU uptake in P4 (a,b) and P6 (c,d) retinas of wild-type (left panels) and TgIRBPE2F1 (right
panels) mice. Cen, central retina; Per, peripheral retina; G, Ganglion cell layer; IPL, inner plexiform layer; PE, pigment epithelium.
Scale bar, 200 mm. (C) Quantitative comparison of BrdU-positive cells in wild-type and TgIRBPE2F1 retinas of dierent ages (P0 ±
P9). BrdU-positive nuclei within a length of 250 mm to optical nerve (center) and to ciliary body (periphery) were counted. The
mean+s.e. is based on data collected from at least three mice
expression of E2F1 may delay certain aspects of retinal We next sought to determine how ectopic expression
development. of E2F1 altered the dierentiation of individual
OncogeneE2F1 and p53 inactivation in retinoblastoma
S-CJ Lin et al
7078
Figure 3 Eects of E2F1 overexpression on retinal apoptosis. (A) Apoptotic cells detected by TUNEL assay in wild-type (left
panels) and TgIRBPE2F1 (right panels) retinas of P4 ± P10 mice. ONL, outer nuclear layer. Scale bar, 200 mm. (B) Quantitative
comparison of apoptotic cells in wild-type and TgIRBPE2F1 retinas. Apoptotic nuclei within a length of 250 mm to optical nerve
(center) and to ciliary body (periphery) were counted. The mean+s.e. is based on data collected from at least three mice
photoreceptors. To accomplish this, opsin protein, but not the peripheral retina in wild-type mice at P2
which is deposited along the lateral cell membrane (data not shown). Cells in the peripheral retina were
and synaptic terminals of the immature rods as shown observed to express opsin at later stages. At P4 and P6,
previously in several other vertebrates (Nir et al., levels of opsin expression appeared to be increased in
1984), was detected by immunohistochemical staining. all rod photoreceptor cells (Figure 4c,e). In transgenic
Opsin ± positive cells were ®rst detected in the central mice, opsin-expressing cells were not seen until P6
OncogeneE2F1 and p53 inactivation in retinoblastoma
S-CJ Lin et al
7079
Figure 4 Eects of E2F1 overexpression on photoreceptors cell dierentiation. (a,b) Formation of OPL in the P6 retinas of wild-
type (a) and TgIRBPE2F1 (b) mice. (c ± f) Expression of opsin in the retinal sections of wild-type (left panel) and TgIRBPE2F1
(right panel) mice. Arrowheads indicate opsin-expressing cells; arrow indicates clusters of apoptotic cells. OPL, outer plexiform
layer. Scale bar, 100 mm
(Figure 4f). At this stage, the opsin expression pattern mice, at which time most regions were depleted of
in the retina of transgenic mice resembles that of the ONL cells, no rosettes were detected (Figure 5f).
P2 retinas in wild-type mice (data not shown). At later Remarkably, there was a striking hemispheric
stages, the photoreceptor cells that are present showed asymmetry with respect to the dysplastic changes and
robust expression of opsin (see below). In summary, the apoptosis discussed above. Divided by the optic
these data indicate that the transgenic expression of nerve, multiple layers of photoreceptor cells were
E2F1 delays normal development in the retina and the arranged into rosette-like structures on one side of
expression of a photoreceptor-speci®c gene, but does the section (Figure 5a), whereas no photoreceptor cells
not prevent eventual photoreceptor dierentiation. were present in the other side (Figure 5c). The loss of
photoreceptor cells was con®rmed by the lack of opsin
staining (Figure 5d). Such initial side-to-side dierence
Dysplasia and degeneration in the neuroretinal cells in
and the ®nal photoreceptor cell loss to the whole retina
TgIRBPE2F1 transgenic mice
suggest that there might be a progressive manifestation
Although the photoreceptors eventually expressed of the E2F1 eects from one retinal hemisphere to the
opsin at apparently normal levels, their dierentiation other; however, the molecular basis for this is not
was still altered in the retinas of transgenic mice. known.
Speci®cally, rosettes were frequently observed in the
outer nuclear layer in the retinas of TgIRBPE2F1 mice,
Loss of p53 promotes both severe dysplasia and
(Figure 5a). These rosettes were composed of at least
hyperplasia in the retina of TgIRBPE2F1 mice but does
partially dierentiated photoreceptors as evidenced by
not prevent photoreceptor degeneration
opsin expression (Figure 5b). The dysplastic lesions
were observed from P9 through P28 TgIRBPE2F1 Because E2F1-mediated cell death has been associated
mice. However, in retinas of 3 month old transgenic with elevated expression of p53 (Hiebert et al., 1995;
OncogeneE2F1 and p53 inactivation in retinoblastoma
S-CJ Lin et al
7080
Figure 5 Asymmetric formation of rosette-like structures and loss of photoreceptor cells in a single TgIRBPE2F1 retina. Retinal
sections from dierent hemispheres of the same TgIRBPE2F1 retina were stained with H&E (a,c,e,f) or immunostained with anti-
opsin antibodies (b,d). (a ± e), TgIRBPE2F1 retinas; (f), wild-type retinas. Ages of the mice are indicated. Arrows indicate the
rosettes. G, Ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer
nuclear layer; OS&IS, outer segment and inner segment; PE, pigment epithelium. Scale bar, (a ± d) 50 mm, (e,f) 100 mm
Kowalik et al., 1998), we tested whether the loss of p53 although it remained signi®cantly higher than in
aects the phenotype of TgIRBPE2F1 mice. TgIRB- normal retinas (data not shown). In 3-week-old and
PE2F1; p537/7 mice were generated after two rounds adult mice, a similar degree of retinal degeneration
of crossings to p53 null mice (Donehower et al., 1992). occurred in TgIRBPE2F1 and TgIRBPE2F1; p537/7
The loss of p53 in the TgIRBPE2F1 mice aected mice (Figure 6c,d). Thus, although the loss of p53
several aspects of the retina phenotype. First, rosette modi®es the degree of apoptosis detectable at any
formation in TgIRBPE2F1; p537/7 mice appeared speci®c time, the loss of photoreceptors in the mature
earlier (from P6 through P14) and dysplastic lesions retina of TgIRBPE2F1 mice is p53-independent. These
were often present in both hemispheres of the retina results indicate that loss of p53 aects multiple aspects
(Figure 6a,b). Second, in some regions of the retina, of the phenotype induced by dysregulated E2F1
multiple layers were not evident. For example, the expression, which may allow excessive hyperplasia
distinct layered structure of ONL in both wild-type and and disorganization of multiple retinal cell types.
TgIRBPE2F1 mice was not observed in TgIRBPE2F1; However, this is still not sucient for oncogenic
p537/7 mice (Figure 6b). At P10, although the OPL transformation of photoreceptors because signi®cant
was evident between the INL and the remaining ONL retinal degeneration still occurs eventually.
in TgIRBPE2F1 mice, the OPL was almost undistin-
guishable in TgIRBPE2F1; p537/7 mice (data not
shown). The INL was also in disarray as evidenced Discussion
by focal expansion to nearly 14-cells thick (Figure
6b,c). The loss of layers in the outer two layers of the E2F transcription factors are important targets for RB-
retina is likely due to focal hyperplasia/dysplasia of dependent control of the cell cycle and tumor
multiple cell types, including photoreceptor cells, formation. We have generated a transgenic mouse
pigment epithelial cells and cells of the INL. Third, model to investigate how the aberrant expression of
the delayed expression of opsin in the TgIRBPE2F1 E2F1 aects photoreceptor cell development and
retina was lessened in the p53 null background. contributes to tumorigenesis. In TgIRBPE2F1 retinas,
Speci®cally, at P6, approximately eight photoreceptors ectopic expression of E2F1 resulted in increased DNA
cells in every row expressed opsin in wild-type retina synthesis and apoptosis in developing photoreceptor
(Figure 4e), yet there was only one opsin-positive cell cells, a ®nding that is similar to what occurs in Rb7/7
in every 5 ± 20 rows in TgIRBPE2F1 retina (Figure 4f). retinas during embryonic development and in retinal
However, there were one or two opsin-positive cells in transplant experiments. The dierentiation of photo-
every three or four rows in TgIRBPE2F1; p537/7 receptor cells, judging from the delayed appearance of
retina (data not shown). Fourth, the level of cell death the OPL and opsin expression, was also impaired but
in the TgIRBPE2F1; p537/7 retinas was much lower not completely blocked. Ultimately, E2F1 deregulation
than that in the TgIRBPE2F1 retinas during P6 to P14, led to photoreceptor cell degeneration rather than
OncogeneE2F1 and p53 inactivation in retinoblastoma
S-CJ Lin et al
7081
Figure 6 Retinal dysplasia and degeneration in TgIRBPE2F1; p537/7 mice. (a ± d) H&E staining of retinal sections of
TgIRBPE2F1; p537/7 mice. Note dysplastic photoreceptor cells showing rosette structures in the P6 (a) and P14 (b) retinas. PE,
pigment epithelium; INL, inner nuclear layer. Scale bar, 100 mm
retinoblastoma formation. p53 de®ciency partially may lead to retinal dysplasia by releasing E2F1 from
rescued the cell death of TgIRBPE2F1 photoreceptor RB (and p107) mediated repression.
cells and promoted retinal transient hyperplasia and Both Rb-de®ciency and E2F1 overexpression alter
dysplasia but did not allow macroscopic tumor opsin expression in the retinal transplant experiments
formation. Together, these studies indicate that normal and transgenic mouse retinas, respectively (Figures 1B
regulation of E2F1, likely by RB-dependent mechan- and 4). These ®ndings are consistent with previous
isms, is required for photoreceptor and retinal studies of how loss of RB alters cellular dierentiation.
development. However, loss of E2F1 regulation is not For example, the trigeminal ganglia of Rb-de®cient
sucient for retinal tumor formation. mice show incomplete dierentiation (Lee et al., 1994).
Several recent studies in other mouse tissues support In Rb heterozygous knockout mice, mutation of the
our conclusions that deregulated E2F1 contributes to remaining wild-type Rb allele alters the dierentiation
the phenotypes associated with Rb inactivation. First, of Rb-null melanotrophs in the pituitary gland. These
aberrant S phase entry and apoptosis in the develop- cells fail to become mature melanotrophs and are not
ing central nervous system and lens of Rb7/7 embryos innervated by growth inhibitory dopaminergic nerve
is suppressed in Rb/E2F1 double mutants (Tsai et al., terminals (Nikitin and Lee, 1996). Finally, in vitro and
1998). Second, mutation of E2F1 suppresses inap- in vivo studies have demonstrated that loss of Rb, or
propriate proliferation and cell death of the lens ®ber ectopic expression of E2F1 inhibits the full expression
cell compartment induced by the HPV E7 oncopro- of genes induced during skeletal muscle and adipose
tein, which binds to and inactivates the RB family dierentiation (Chen and Lee, 1999; Chen et al., 1996;
proteins (McCarey et al., 1999). In these respects, the Classon et al., 2000; Novitch et al., 1996; Zacksenhaus
eects of aberrant expression of E2F1 in mouse et al., 1996). Taken together, these data indicate that
photoreceptors are similar to eects in the lens and loss of RB function and ectopic expression of E2F1 is
other areas of the central nervous system. Further- sucient to prevent normal dierentiation of numerous
more, our transgenic mouse model adds further types of cells. Interestingly, it does not completely
insight into how RB and the RB-related protein prevent dierentiation per se but rather it appears to
p107 aects retinal development. Previous studies have delay the expression of a dierentiation-speci®c gene in
shown that knock out mice and chimeric mice in photoreceptors. Although photoreceptors in our trans-
which all or some of the retina is derived from Rb+/ genic mice eventually show robust opsin expression, we
7
; p1077/7 or Rb7/7; p1077/7 cells have dysplasia have not performed a quantitative assay to compare
(Lee et al., 1996; Robanus-Maandag et al., 1998) that the level of expression with normal photoreceptors. At
is similar to what we observed in our studies of E2F1. a molecular level, how loss of Rb alters dierentiation
Thus, our data suggest that loss of the Rb and p107 is not known. It has been shown to interact with a
OncogeneE2F1 and p53 inactivation in retinoblastoma
S-CJ Lin et al
7082
number of transcription factors that mediate cellular cell proliferation of other retinal cells in a p53-
dierentiation (Chen et al., 1996; Gu et al., 1993; dependent manner. However, the molecular mechan-
Lipinski and Jacks, 1999; Schneider et al., 1994). On isms by which this may occur are not known. It is also
the other hand, it is conceivable that loss of Rb plausible that there might be low levels of ectopic E2F1
indirectly alters the activity of transcription factors that expression in non-photoreceptor cells and the resultant
drive cellular dierentiation by releasing E2F activity growth promoting eect is detectable only in the
(Wang et al., 1995). Finally, it is also conceivable that absence of p53. Further experiments will be required
either loss of Rb or aberrant E2F1 expression could to clarify the mechanisms involved.
in¯uence cellular dierentiation by allowing for the One notable dierence between our studies and
accumulation of additional genetic changes. Further previous studies of the RB gene pathway in the retina
experiments will be required to clarify the molecular is the development of macroscopic retinal tumors,
basis by which loss of Rb or aberrant expression of which occurred either in Rb7/7; p1077/7 chimeric mice
E2F1 blocks the normal dierentiation of photorecep- (Robanus-Maandag et al., 1998) or in the TgIRBPE7;
tor cells. p537/7 mice (Howes et al., 1994). The molecular basis
Compared to other models in which E2F1 is for the transient dysplasia and hyperplasia that occurs
ectopically expressed, our study of E2F1-expressing in our TgIRBPE2F1; p537/7 mice is not known. It is
photoreceptor cells oers several new observations. certainly conceivable that pro-apoptotic mechanisms in
First, hyperplasia was not observed in photoreceptors the retina are activated in the presence of ectopically
in our mice whereas transgenic expression of E2F-1 in expressed E2F1 to prevent tumor formation in the
squamous epithelial cells caused increased cell prolif- retina. In this regard, it is interesting to note that
eration and hyperplasia despite increased apoptosis overexpression of SV40 T antigen under the control of
(Pierce et al., 1998a). Second, the loss of p53 only the rhodopsin promoter also results in retinal degen-
partially abrogates E2F1-mediated cell death and only eration in mice (al-Ubaidi et al., 1992). However, these
transiently sustains dysplasia and hyperplasia in photoreceptor cells proliferate in tissue culture and are
photoreceptors of our TgIRBPE2F1 mice (Figure 6), tumorigenic when implanted subcutaneously into nude
while E2F1-induced apoptosis in the epidermal cells is mice (al-Ubaidi et al., 1992). Furthermore, in the retina
largely p53-dependent and its loss allows for the of Rb7/7 chimeric mice, there was severe cellular
formation of spontaneous skin tumors (Pierce et al., degeneration during E16.5 ± E18.5 (Maandag et al.,
1998b). Similarly, apoptosis in Rb-de®cient embryos 1994). The contribution of Rb7/7 retinal cells was
depends on p53 and E2F1 in the central nervous reduced to less than 15% in adult mice (Maandag et
system (Macleod et al., 1996; Tsai et al., 1998). Finally, al., 1994). Similarly, our intracranial Rb7/7 retinal
we demonstrated that the forced expression of E2F1 transplants exhibited signi®cant apoptosis 1 week after
alters photoreceptor and retina development (Figures 4 transplantation, a time point equivalent to E18.5 of the
and 5); however, similar forced expression of E2F1 in donor tissue (Figure 1B). However, transplanted Rb7/7
epidermal cells does not appear to block cellular retinal cells were able to survive for more than 2
dierentiation (Pierce et al., 1998a). In summary, these months (data not shown). Taken together, these results
®ndings indicate that the eects of deregulated RB/ indicate the presence of apoptotic factors in situ in the
E2F1 on cellular proliferation, dierentiation, and eyes of the transgenic mice. It is conceivable that these
apoptosis dier in speci®c types of cell and tissue. pro-apoptotic factors are disrupted in the TgIRB-
Although no macroscopic tumors form in TgIRB- PE7; p537/7 mice but not in our TgIRBPE2F1; p537/7
PE2F1; p537/7 mice, there is transient hyperplasia and mice. Delineating mechanisms underlying phenotypic
dysplasia of multiple retinal cell types (Figure 6a,b). dierences between genetic loss of Rb and p107 (Lee et
This unexpected ®nding suggests that aberrant expres- al., 1996; Robanus-Maandag et al., 1998) or by
sion of E2F1 in photoreceptors may promote pro- expression of IRBP-E7 in the p537/7 background
liferation of nearby p53-de®cient inner retinal cells and (Howes et al., 1994), versus the non-tumorigenic
pigment epithelial cells through a paracrine mechan- properties of IRBP ± E2F1 (even in the absence of
ism. There is a precedent for paracrine regulation of p53) would likely lead to better understanding of the
cells in vertebrate retinas. For example, during functions of these gene products and the biology of
development, the seven major types of cells in the human retinoblastoma.
retina are derived from a pool of multipotent
progenitor cells (Cepko et al., 1996). Numerous studies
have shown that cell ± cell interactions are involved in
cell fate determination during this time (Belliveau and Materials and methods
Cepko, 1999; Belliveau et al., 2000; Cepko et al., 1996).
Similarly, studies using intact frog retina indicate that Construction of TgIRBPE2F1 transgene and generation of
the phosphorylation status of rhodopsin is in part transgenic mice
regulated by dopamine released from the inner cell A plasmid pBSpKCR3 modi®ed from pKCR3 that consists of a
layers (Udovichenko et al., 1998). Clearly this is minor 1.9 kb fragment of the murine IRBP promoter, a 1.2 kb part of
compared to the phosphorylation induced by light. the rabbit b globin gene including partial exon 2, intron 2, and
Thus, it seems reasonable to speculate that the aberrant exon 3, and a polyadenylation site, was provided by Dr Jolene
expression of E2F1 in photoreceptors could alter the Windle (Howes et al., 1994). A 1.5 kb fragment containing the
OncogeneE2F1 and p53 inactivation in retinoblastoma
S-CJ Lin et al
7083
full-length human E2F1 cDNA was inserted into exon 3 of the in formalin, and embedded in paran. Tissue sections were
rabbit b globin gene of pBSpKCR3. The TgIRBPE2F1 treated with 4N HCl/0.2% Triton X-100, and processed for
transgene was cut with KpnI and XbaI, gel-puri®ed, and immunostaining as described above using anti-BrdU anti-
microinjected into the male pronucleus of fertilized eggs bodies (1 : 200, Becton Dickinson). The terminal deoxyribo-
derived from CB6 F16C57BL/6 intercrosses. Transgenic nucleotidyltransferase-mediated dUTP-biotin nick end
founder mice were identi®ed by Southern blot analysis of tail- labeling (TUNEL) assay was performed as described (Howes
derived genomic DNA. Transgenic mice of subsequent et al., 1994).
generations were identi®ed by PCR using primers derived from
the human E2F1 cDNA sequence, 5' primer (5'-GGAAACT-
Quantification of cells in the neuroretina
GACCATCAGTACCTG-3'), and 3' primer (5'-CCAGCCAC-
TGGATGTGGTTCTT-3'). Transgenic lines were established Quanti®cation of the number of cells positive for BrdU and
by breeding founders to C57BL/6 mice. Subsequent genera- TUNEL was performed using light microscopy. The numbers
tions of transgenic mice were maintained on a mixed CB6 of nuclei in the neuroretina within a length of 250 mm to the
F16C57BL/6 genetic background. optical nerve (the central region) and to the ciliary body (the
peripheral region) were counted. At least three mice were
examined to obtain the mean and the standard error.
Histological analysis
Samples were ®xed in 10% buered formalin and processed
Retinal transplantation methods
through paran embedding follow standard procedures.
Sections (4 mm thick) were cut parallel to the optic nerve Embryos were obtained from anesthetized, timed-pregnant
and stained with hematoxylin and eosin (H&E) for light heterozygous Rb-1D20/+ mice (Lee et al., 1992). Retinas were
microscopy. removed from E11.5 ± E12.5 embryos and transplanted into
the superior colliculus or cerebral cortex of 1-day-old (P1)
wild-type or Rb-1D20/+ neonates as described (Hankin and
E2F1 and opsin immunostaining
Lund, 1987). Brie¯y, a small glass pipet containing the donor
Immunostaining was performed using the Vectastain Elite tissue and sterile F10 culture medium (GIBCO BRL) was
ABC Kit (Vector Laboratories). Brie¯y, paran-embedded inserted through a small hole in the cartilagenous skull
sections were rehydrated, ®xed in methanol containing 3% overlying the recipient midbrain or cortex. The donor tissue
hydrogen peroxide for 5 min, and blocked in 1% normal was ejected from the pipet with 1 ml of F10.
horse serum (NHS) in PBS for 20 min. The sections were
incubated with anti-E2F1 monoclonal antibodies (1 : 200,
Pharmingen) or mouse anti-opsin antibodies (1 : 2000) diluted
in 2% NHS/PBS overnight at 48C. After washes in PBS,
sections were incubated with biotinylated anti-mouse IgG for
30 min. Slides were washed in PBS, incubated with Acknowledgments
streptavidin/biotinylated peroxidase conjugate, developed by We thank Dr Jolene Windle for the plasmid pBSKCR3,
Vector VIP or DAB substrates (Vector Laboratories), and Nancy Ransom for technical advice and Dr CL Cepko for
counterstained with methyl green or hematoxylin. helpful comment. We appreciate Drs NP Hu's, Y Min's,
and E Gerbino's contribution in the early phase of the
project. We also wish to thank K Bushnell for proof-
BrdU incorporation and TUNEL assays
reading the manuscript. This work was supported by NIH
Mice received bromodeoxyuridine (BrdU) at a dose of grants CA49649 to E Lee. Histology was performed in part
100 mg/g of body weight by intraperitoneal and subcutaneous by a core facility of the San Antonio Cancer Institute
injection. After 2 h, retina tissue samples were collected, ®xed (grant #P30 CA 54174).
References
Adams PD and Kaelin Jr WG. (1995). Semin. Cancer Biol., 6, Classon M, Kennedy BK, Mulloy R and Harlow E. (2000).
99 ± 108. Proc. Natl. Acad. Sci. USA, 97, 10826 ± 10831.
al-Ubaidi MR, Holly®eld JG, Overbeek PA and Baehr W. DeGregori J, Leone G, Miron A, Jakoi L and Nevins JR.
(1992). Proc. Natl. Acad. Sci. USA, 89, 1194 ± 1198. (1997). Proc. Natl. Acad. Sci. USA, 94, 7245 ± 7250.
Belliveau MJ and Cepko CL. (1999). Development, 126, Donehower LA, Harvey M, Slagle BL, McArthur MJ,
555 ± 566. Montgomery Jr CA, Butel JS and Bradley A. (1992).
Belliveau MJ, Young TL and Cepko CL. (2000). J. Neurosci., Nature, 356, 215 ± 221.
20, 2247 ± 2254. Duronio RJ, O'Farrell PH, Xie JE, Brook A and Dyson N.
Brehm A and Kouzarides T. (1999). Trends Biochem. Sci., 24, (1995). Genes Dev., 9, 1445 ± 1455.
142 ± 145. Dyson N. (1998). Genes Dev., 12, 2245 ± 2262.
Cepko CL, Austin CP, Yang X, Alexiades M and Ezzeddine Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin Jr WG,
D. (1996). Proc. Natl. Acad. Sci. USA, 93, 589 ± 595. Livingston DM, Orkin SH and Greenberg ME. (1996).
Chen G and Lee EY. (1999). DNA Cell Biol., 18, 305 ± 314. Cell, 85, 549 ± 561.
Chen PL, Riley DJ, Chen Y and Lee WH. (1996). Genes Dev., Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V
10, 2794 ± 2804. and Nadal-Ginard B. (1993). Cell, 72, 309 ± 324.
Clarke AR, Maandag ER, van Roon M, van der Lugt NM, Hankin MH and Lund RD. (1987). J. Comp. Neurol., 263,
van der Valk M, Hooper ML, Berns A and te Riele H. 455 ± 466.
(1992). Nature, 359, 328 ± 330.
OncogeneE2F1 and p53 inactivation in retinoblastoma
S-CJ Lin et al
7084
Hankin M, Sefton AJ and Lund RD. (1993). Brain Res. Dev. Nir I, Cohen D and Papermaster DS. (1984). J. Cell Biol., 98,
Brain Res., 75, 146 ± 150. 1788 ± 1795.
Hiebert SW, Packham G, Strom DK, Haner R, Oren M, Novitch BG, Mulligan GJ, Jacks T and Lassar AB. (1996).
Zambetti G and Cleveland JL. (1995). Mol. Cell Biol., 15, J. Cell Biol., 135, 441 ± 456.
6864 ± 6874. Pierce AM, Fisher SM, Conti CJ and Johnson DG. (1998a).
Howes KA, Ransom N, Papermaster DS, Lasudry JG, Oncogene, 16, 1267 ± 1276.
Albert DM and Windle JJ. (1994). Genes Dev., 8, 1300 ± Pierce AM, Gimenez-Conti IB, Schneider-Broussard R,
1310. Martinez LA, Conti CJ and Johnson DG. (1998b). Proc.
Hu N, Gutsmann A, Herbert DC, Bradley A, Lee WH and Natl. Acad. Sci. USA, 95, 8858 ± 8863.
Lee EY. (1994). Oncogene, 9, 1021 ± 1027. Qin XQ, Livingston DM, Kaelin Jr WG and Adams PD.
Ishizaki J, Nevins JR and Sullenger BA. (1996). Nat. Med., 2, (1994). Proc. Natl. Acad. Sci. USA, 91, 10918 ± 10922.
1386 ± 1389. Robanus-Maandag E, Dekker M, van der Valk M, Carrozza
Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA ML, Jeanny JC, Dannenberg JH, Berns A and te Riele H.
and Weinberg RA. (1992). Nature, 359, 295 ± 300. (1998). Genes Dev., 12, 1599 ± 1609.
Johnson DG, Cress WD, Jakoi L and Nevins JR. (1994). Ross JF, Liu X and Dynlacht BD. (1999). Mol. Cell, 3, 195 ±
Proc. Natl. Acad. Sci. USA, 91, 12823 ± 12827. 205.
Johnson DG, Schwarz JK, Cress WD and Nevins JR. (1993). Schneider JW, Gu W, Zhu L, Mahdavi V and Nadal-Ginard
Nature, 365, 349 ± 352. B. (1994). Science, 264, 1467 ± 1471.
Kowalik TF, DeGregori J, Leone G, Jakoi L and Nevins JR. Shan B and Lee WH. (1994). Mol. Cell Biol., 14, 8166 ± 8173.
(1998). Cell Growth Dier., 359, 113 ± 118. Singh P, Wong SH and Hong W. (1994). EMBO J., 13,
Kowalik TF, DeGregori J, Schwarz JK and Nevins JR. 3329 ± 3338.
(1995). J. Virol., 69, 2491 ± 2500. Skapek SX, Jansen D, Wei TF, McDermott T, Huang W,
Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Olson EN and Lee EY. (2000). J. Biol. Chem., 275, 7212 ±
Lee WH and Bradley A. (1992). Nature, 359, 288 ± 294. 7223.
Lee EY, Hu N, Yuan SS, Cox LA, Braley A, Lee WH and Slansky JE and Farnham PJ. (1996). Curr. Top Microbiol.
Herrup K. (1994). Genes Dev., 8, 2008 ± 2021. Immunol., 208, 1 ± 30.
Lee MH, Williams BO, Mulligan G, Mukai S, Bronson RT, Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L and
Dyson N, Harlow E and Jacks T. (1996). Genes Dev., 10, Jacks T. (1998). Mol. Cell., 2, 293 ± 304.
1621 ± 1632. Udovichenko IP, Newton AC and Williams DS. (1998). J.
Lin SC, Skapek SX and Lee EY. (1996). Semin. Cancer Biol., Biol. Chem., 273, 7181 ± 7184.
7, 279 ± 289. Wang J, Helin K, Jin P and Nadal-Ginard B. (1995). Cell
Liou GI, Geng L, al-Ubaidi MR, Matragoon S, Hanten G, Growth Dier., 6, 1299 ± 1306.
Baehr W and Overbeek PA. (1990). J. Biol. Chem., 265, Wu CL, Classon M, Dyson N and Harlow E. (1996). Mol.
8373 ± 8376. Cell Biol., 16, 3698 ± 3706.
Lipinski MM and Jacks T. (1999). Oncogene, 18, 7873 ± 7882. Wu X and Levine AJ. (1994). Proc. Natl. Acad. Sci. USA, 91,
Maandag EC, van der Valk M, Vlaar M, Feltkamp C, 3602 ± 3606.
O'Brien J, van Roon M, van der Lugt N, Berns A and te Xu G, Livingston DM and Krek W. (1995). Proc. Natl. Acad.
Riele H. (1994). EMBO J., 13, 4260 ± 4268. Sci. USA, 92, 1357 ± 1361.
Macleod KF, Hu Y and Jacks T. (1996). EMBO J., 15, Yamasaki L, Bronson R, Williams BO, Dyson NJ, Harlow E
6178 ± 6188. and Jacks T. (1998). Nat. Genet., 18, 360 ± 364.
McCarey J, Yamasaki L, Dyson NJ, Harlow E and Griep Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E and
AE. (1999). Mol. Cell. Biol., 19, 6458 ± 6468. Dyson NJ. (1996). Cell, 85, 537 ± 548.
Morgenbesser SD, Williams BO, Jacks T and DePinho RA. Young RW. (1984). Cell, 85, 537 ± 548.
(1994). Nature, 371, 72 ± 74. Young RW. (1985). Anat. Rec., 212, 199 ± 205.
Mulligan G and Jacks T. (1998). Trends Genet., 14, 223 ± 229. Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA
Nevins JR. (1998). Cell Growth Dier., 9, 585 ± 593. and Gallie BL. (1996). Genes Dev., 10, 3051 ± 3064.
Nikitin A and Lee WH. (1996). Genes Dev., 10, 1870 ± 1879. Zhang HS, Postigo AA and Dean DC. (1999). Cell, 97, 53 ± 61.
OncogeneYou can also read

















































