A Method for Rapid Screening, Expression, and Purification of Antimicrobial Peptides
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
microorganisms
Article
A Method for Rapid Screening, Expression, and Purification of
Antimicrobial Peptides
Yingli Zhang 1 , Zhongchen Li 1 , Li Li 1 , Ben Rao 2 , Lixin Ma 1 and Yaping Wang 1, *
1 State Key Laboratory of Biocatalysis and Enzyme, Engineering Hubei Collaborative Innovation Center for
Green Transformation of Bio-Resources, Hubei Key Laboratory of Industrial Biotechnology,
Biology Faculty of Hubei University, Hubei University, Wuhan 430062, China; zyl19980703@163.com (Y.Z.);
lizc0408@163.com (Z.L.); groundcontrol2021@163.com (L.L.); malixing@hubu.edu.cn (L.M.)
2 National Biopesticide Engineering Technology Research Center, Hubei Biopesticide Engineering Research
Center, Hubei Academy of Agricultural Sciences, Biopesticide Branch of Hubei Innovation Centre of
Agricultural Science and Technology, Wuhan 430064, China; raoben1983729@aliyun.com
* Correspondence: stwangyaping@163.com
Abstract: In this study, a method for the rapid screening, expression and purification of antimicrobial
peptides (AMPs) was developed. AMP genes were fused to a heat-resistant CL7 tag using the SLOPE
method, and cloned into Escherichia coli and Pichia pastoris expression vectors. Twenty E. coli and ten
P. pastoris expression vectors were constructed. Expression supernatants were heated, heteroproteins
were removed, and fusion proteins were purified by nickel affinity (Ni-NTA) chromatography. Fusion
proteins were digested on the column using human rhinovirus (HRV) 3C protease, and AMPs were
released and further purified. Five AMPs (1, 2, 6, 13, 16) were purified using the E. coli expression
system, and one AMP (13) was purified using the P. pastoris expression system. Inhibition zone
and minimum inhibitory concentration (MIC) tests confirmed that one P. pastoris¬-derived and two
Citation: Zhang, Y.; Li, Z.; Li, L.; Rao,
E. coli-derived AMPs have the inhibition activity. The MIC of AMP 13 and 16 from E. coli was 24.2 µM,
B.; Ma, L.; Wang, Y. A Method for
and the MIC of AMP 13 from P. pastoris was 8.1 µM. The combination of prokaryotic and eukaryotic
Rapid Screening, Expression, and
expression systems expands the universality of the developed method, facilitating screening of a
Purification of Antimicrobial
Peptides. Microorganisms 2021, 9, 1858.
large number of biologically active AMPs, establishing an AMP library, and producing AMPs by
https://doi.org/10.3390/ industrialised biological methods.
microorganisms9091858
Keywords: antimicrobial peptides expression; CL7 tag; Escherichia coli; Pichia pastoris
Academic Editor:
Giuseppantonio Maisetta
Received: 23 July 2021 1. Introduction
Accepted: 22 August 2021
Antimicrobial peptides (AMPs) are small, naturally occurring peptides 5–100 amino
Published: 1 September 2021
acids in length, and possess high structural diversity [1–3]. Structures include α-helices
and β-sheet regions rich in cysteine, and regions containing a high percentage of spe-
Publisher’s Note: MDPI stays neutral
cific amino acids with unique structures. AMPs are produced in various eukaryotic and
with regard to jurisdictional claims in
prokaryotic organisms, and some affect the innate immune systems of bacteria, plants,
published maps and institutional affil-
insects and mammals.
iations.
Recently, there has been a surge in the use of small peptides as drug candidates because
they provide significant advantages over traditional small molecule drugs. AMPs have the
advantages of low molecular weight, high heat resistance, broad-spectrum antimicrobial
activity and, importantly, they show minimal antimicrobial resistance, making them a
Copyright: © 2021 by the authors.
promising treatment for antimicrobial therapy [4].
Licensee MDPI, Basel, Switzerland.
AMPs protect against a wide variety of pathogens, including prokaryotic microorgan-
This article is an open access article
isms, fungi, viruses, protozoa, insect cells and cancer cells. Advantages include antibacterial
distributed under the terms and
activity, degradability in vivo and, because they are from natural sources, they are gener-
conditions of the Creative Commons
ally safe and have non-toxic effects and, hence, are applicable in foods and feed additives.
Attribution (CC BY) license (https://
However, there are also some limitations for AMP applications. The natural AMPs are
creativecommons.org/licenses/by/
4.0/).
labile, depending on the surrounding environments, such as the presence of protease, pH
Microorganisms 2021, 9, 1858. https://doi.org/10.3390/microorganisms9091858 https://www.mdpi.com/journal/microorganisms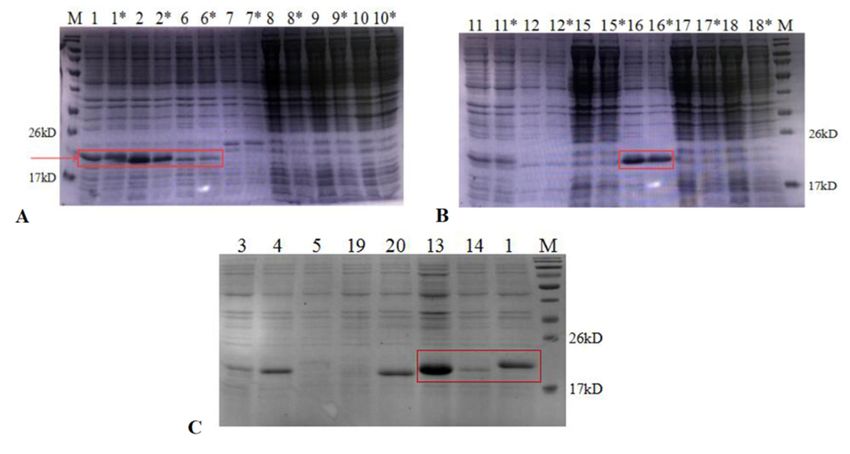
Microorganisms 2021, 9, 1858 2 of 15
change, and so on. The potential toxicity of AMPs for oral application and the high cost of
peptide production are the other obstacles [5,6].
There are various ways to obtain AMPs [7–12]: (1) They can be obtained directly from
animals and plants, but this has the disadvantages of high cost, low yield, cumbersome
technical operations and often insufficient purity. (2) They can be prepared by chemi-
cal synthesis, such as solid-phase synthesis, fragment condensation and other synthetic
peptide procedures. Chemically synthesised AMPs are generally of good purity and are
suitable for the production of high-value pharmacological-grade peptides, but they have
disadvantages including expensive synthesis and low chemical reaction efficiency. (3) Enzy-
matic hydrolysis using proteases can yield AMPs with higher activity than other methods.
However, since the target sequence or structure is not artificially designed, purification may
be difficult and costly. (4) Finally, genetic engineering technology can be used to produce
AMPs, and this may eventually become the main method.
Both eukaryotic and prokaryotic systems are currently used for heterologous expres-
sion of AMPs [13]. The characteristics of various AMPs, including structure, disulfide
bond abundance, acidic/basic residue distribution, isoelectric point and degree toxicity, all
place different demands on the requirements for host selection. The most commonly used
expression engineering strains are Escherichia coli, Bacillus subtilis and Pichia pastoris. In the
past few decades, researchers have used several expression systems to produce AMPs in
a cost-effective manner, and E. coli alone has been used to produce more than 80% of all
recombinant AMPs. However, E. coli cells are not suitable for small peptides expressed at
high concentrations, and the toxicity of AMPs to the host cannot be ignored.
The P. pastoris yeast strain has been developed into an excellent engineered host for
high-concentration and high-purity heterologous expression of proteins, including short
peptides from different sources [14,15]. This host has been successfully used to produce a
variety of functional recombinant proteins from humans, animals, plants, fungi, bacteria
and viruses. Most importantly, this host can express recombinant AMPs in secreted form,
which provides several advantages over prokaryotic expression systems, including correct
folding in the cell, formation of disulfide bonds, correct protein tertiary structure and other
post-translational modifications that affect protein function. The secretion of heterologous
proteins prevents their accumulation from affecting the normal growth of the host, and
avoids intracellular protein contamination. In addition, extracellular secretion simplifies the
purification steps, and high-density fermentation enables industrial production of AMPs.
Due to these advantages of the P. pastoris expression system, it has become important in
the industrial production of AMPs.
In this study, we developed a method for rapid screening, expression and purification
of AMPs. We synthesised small peptides using the SLOPE method, which greatly improves
the efficiency of constructing AMP expression vectors, and has lower costs. AMP genes
were fused to a heat-resistant CL7 tag [16,17] using the SLOPE method, and cloned into
Escherichia coli and Pichia pastoris expression vectors. Twenty E. coli and ten P. pastoris
expression recombinants were constructed. The CL7 tag promotes solubilisation, good ther-
mal stability and provides heat resistance to the fusion protein. Expression supernatants
were heated, heteroproteins were removed, and fusion proteins were purified by nickel
affinity (Ni-NTA) chromatography. Fusion proteins were digested on the column using
high-efficiency low-cost human rhinovirus (HRV) 3C protease, and AMPs were released
and further purified. Five AMPs (1, 2, 6, 13, 16) were screened and purified using the
E. coli expression system, and one AMP (13) was purified using the P. pastoris expression
system. Inhibition zone and minimum inhibitory concentration (MIC) tests confirmed that
one P. pastoris-derived and two E. coli-derived AMPs have the inhibition activity. The MIC
of AMP 13 and 16 from E. coli was 24.2 µM, and the MIC of AMP 13 from P. pastoris was
8.1 µM. The combination of prokaryotic and eukaryotic expression systems expands the
universality of the developed method, facilitating screening of a large number of biologi-
cally active AMPs, establishing an AMP library and producing AMPs by industrialised
biological methods.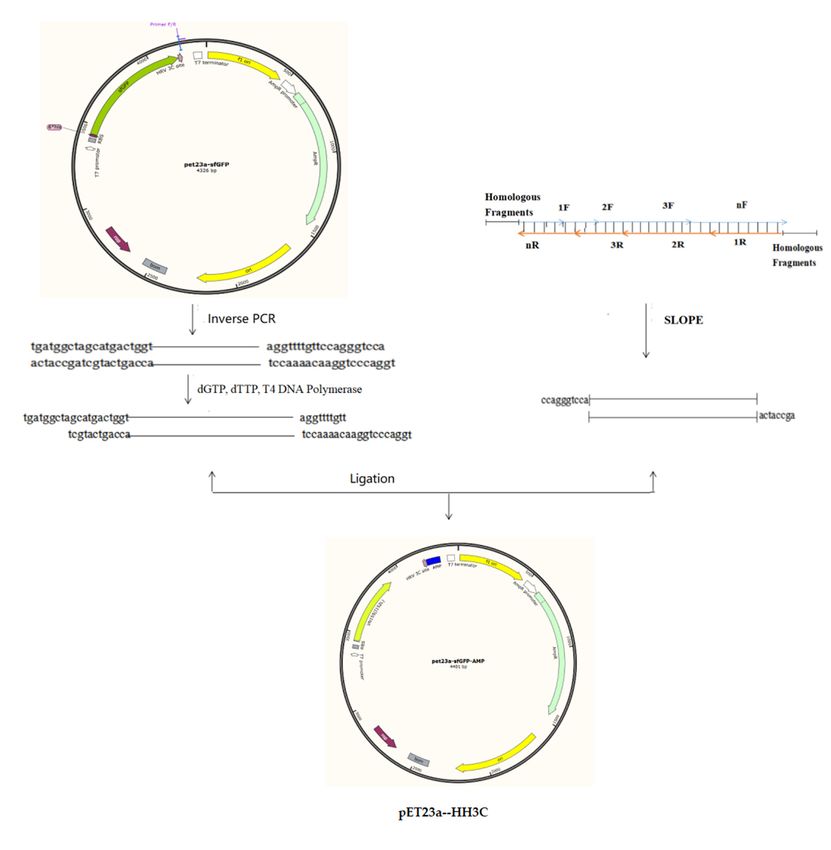
Microorganisms 2021, 9, 1858 3 of 15
2. Materials and Methods
2.1. Strains, Reagents and Media
Pichia pastoris GS115 and E. coli DH5α/BL21 strains were purchased from Invitrogen
(Shanghai, China). The pHBM905BDM plasmid was constructed in our laboratory. All
culture media, including Luria–Bertani (LB), minimal dextrose (MD), buffered minimal
glycerol (BMGY) and buffered minimal methanol (BMMY) were prepared as described in
the P. pastoris expression manual (Invitrogen).
2.2. Construction of the E. coli Expression Vector pET23a-HH3C
The starting vector was pET23a. A fusion gene containing a 6× HIS sequence (18 bp), a
CL7 sequence (390 bp), and a human rhinovirus (HRV) 3C sequence (24 bp) was synthesised
by Sangon BioTech (Shanghai, China). This fusion gene was inserted into the pET23a vector
according to the method described by Li et al. [18], resulting in plasmid pET23a–HH3C.
Using the same method, the fusion gene was inserted into the pPicZα and pHBM905BDM
vectors, resulting in pPICZα-HH3C and pHBM905BDM-HH3C, respectively.
2.3. Synthesis of AMP Nucleotide Sequences
We selected AMP amino acid sequences from the AMP library website (http://aps.
unmc.edu/AP/main.php (accessed on 16 October 2018)). The nucleotide sequences of
these genes were optimised according to the codon preferences of E. coli and P. pastoris. We
designed several pairs (2, 3, 4) of primers according to the AMP nucleotide sequence. These
primers were then mixed, and a SLOPE reaction was performed as follows: 98 ◦ C for 3min,
37 ◦ C for 30min, 12 ◦ C for 10min. After SLOPE reactions were performed, the products
were used as templates. The universal vector pET23a-HH3C was amplified by inverse PCR
to obtain the linearising vector [19,20]. Then, T4 DNA polymerase was utilized to treat
the vector to obtain vector fragments with sticky ends. The AMP primers were equipped
with adapters that are complementary to the sticky end of the vector. Importantly, each
primer only needs to go through a single SLOPE reaction to obtain the product of the gene
fragment to be used. The vector fragment treated with T4 polymerase and the SLOPE
reaction product were mixed at a molar ratio of 1:3, and the ligated products were used to
transform E. coli cells. The resulting plasmids were confirmed by PCR and sequencing.
2.4. Expression of AMPs in E. coli
The plasmids pET23a-HH3C-AMP were transformed into E. coli BL21. The recombi-
nant strains were induced by IPTG. After induction, bacteria were collected in a 50 mM
centrifuge tube, centrifuged at 9000 rpm for 10 min, and the supernatant was discarded.
After adding 30 mM HRV 3C Cleavage Buffer (50 mM Tris, 150 mM NaCl, pH7.5) to
resuspend cells, they were centrifuged, and this step was repeated once or twice. After
washing, the cell pellet from 100 mL bacterial solution was resuspended in 20 mM HRV
3C Cleavage Buffer to prepare for ultrasonic cell disruption. Bacteria were lysed until
the solution was clear, and samples were centrifuged at high speed (≥10,000 rpm) for
10 min. After centrifugation, the supernatant was subjected to SDS-PAGE to assess protein
expression. In addition, as a control, the same volume (1 mL) of each bacterial supernatant
was heated to 80 ◦ C for 30 min. After high temperature treatment, the supernatant was
centrifuged at high speed and the supernatant was collected for SDS-PAGE.
2.5. HRV 3C Cleavage
The nickel column was activated by HRV 3C Cleavage Buffer containing 10 mM
imidazole, and then the protein sample was subjected to the column, which was washed
three times with HRV 3C Cleavage Buffer, with 3 bed volumes each time. HRV 3C Cleavage
Buffer was added to resuspend the nickel beads at a bed volume:reaction buffer (2ml HRV
3C Cleavage Buffer, 100 µL 100mM EDTA, 5 µL HRV 3C protease) volume ratio of 1:1.
Digestion was performed at 4 ◦ C on a slow shaker for 3 to 4 h. After digestion, the column
was eluted with one bed volume of 1 M NaCl, and the flow-through was collected which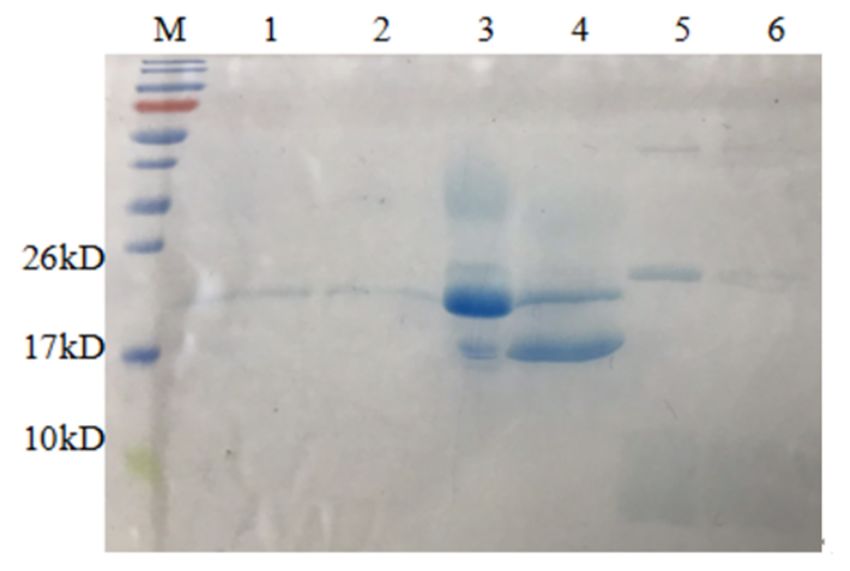
Microorganisms 2021, 9, 1858 4 of 15
was subjected to the following experiments. Then, the AMP concentrations were measured
by the Bradford method.
2.6. Inhibition Testing
To measure AMP activity, the agarose plate method was employed. Microorganisms
for inhibition testing were activated, 3–5 single colonies were picked and placed in liquid
LB medium, cultured on a shaker at 37 ◦ C for 6 h. Solid LB medium was melted by
heating, 500 µL of liquid bacterial culture was added for every 100 mL of medium after
cooling slightly, and the mixture was poured into a bacterial plate to solidify. Oxford
cups were inserted into the solidified mixed bacterial plate, while being careful not to
penetrate the medium completely, so as not to affect the penetration of the test liquid on the
plate. Attention was paid to the spacing between the Oxford cups. Corresponding Oxford
cups were marked and positive/negative controls (100 µL each) were added. Plates were
incubated at 37 ◦ C overnight to observe the antibacterial effects.
2.7. MIC Testing
When measuring the minimum inhibitory concentration (MIC) of the expressed and
purified AMPs, we performed the test according to the Clinical and Laboratory Standards
Association. Specifically, microorganisms to be tested were activated, and five colonies
Microorganisms 2021, 9, x FOR PEER REVIEW 6 of 16
were picked and separately cultured into liquid medium on a shaker (220 rpm) at 37 ◦ C
until the absorbance at 600 nm (OD600 ) reached 0.13–0.17; thus, corresponding to a biomass
concentration of ~1 × 108 colony-forming units (CFU)/mL. Samples for testing were
diluted to 4 mM, and two-fold serial GGTCTTGTGCGCAAAGGAGGTGAGAAGTTT-
dilutions were performed in a 96-well plate, with
GLVRKGGEKFGEKLRKIGQKIKEFFQKL
the concentration ranging from 100 to 0.195 GGCGAAAAGTTACGCAAAATCGGG-
µM. Each dilution (50 µL) was performed in
AP00498(AMP15)
ALEIEQ(3.95)
duplicate. culture containing ~1 × 106 CFU/mL was added to
Next, 50 µL of microbial CAAAAAATCAAGGAGTTTTTTCAAAAATTAGCT
each well. After incubating overnight at 37 ◦ C, OD 600 was measured using a microplate
CTGGAGATTGAACAA
spectrophotometer. The MIC is definedGACTCCCATGCTAAGAGACACCACGGC-
as the minimum test compound concentration
(µM)DSHAKRHHGYKRK-
required to visibly inhibit the growth of microorganisms.
AP00505(AMP16) TATAAACGTAAGTTTCACGAGAAGCAC-
FHEKHHSHRGY(3.04)
CATTCTCACCGTGGATAT
3. Results
TTTCTGGGGGGCCTTATCAAGATCGTAC-
AP00513(AMP17) 3.1. Synthesis of AMP DNA Sequences Using SLOPE Technology
FLGGLIKIVPAMICAVTKKC(2.11)
CAGCCATGATATGCGCCGTCACGAAGAAATGT
We used SLOPE technology to synthesise the nucleotide sequences of AMPs. SLOPE
ATATGGTTGACGGCTCTTAAATTTCTGG-
IWL-
is used for the annealing of multiple primers to synthesise small fragments. This method
AP00516(AMP18) GAAAACACGCTGCGAAGCACCTT-
TALKFLGKHAAKHLAKQQLSKL(2.85)
avoids the disadvantages of expensive gene synthesis and the low efficiency of PCR
GCGAAACAGCAGCTTAGCAAACTT
amplification. It is suitable for small fragment molecular cloning.
CAATGGGGAAGAA-
We chose twenty AMPs (Table 1) from the antimicrobial peptide library website for
AP00519(AMP19) QWGRRCCGWGPGRRYCRRWC(2.54) GATGCTGCGGCTGGGGTCCTGGACGCAGA-
production using the E. coli expression system. The DNA sequences of these AMPs were
synthesised according to codon preference. TheTACTGCCGCCGTTGGTGT
PCR results showed that the size of the
TGGCTGAATGCCTTATTACACCAC-
AMP fragments obtained by the SLOPE reaction were consistent with the theoretical sizes
AP00538(AMP20) WLNALLHHGLNCAKGVLA(1.93)
of the corresponding AMPs (Figure 1).GGTCTGAACTGTGCCAAAGGTGTACTGGCC
Figure1.1.Agarose
Figure Agarosegel
gel(1.5%)
(1.5%)analysis
analysisof
ofSLOPE
SLOPEproducts
productsof
ofAMPs.
AMPs.M,
M,DL2000
DL2000DNA
DNAmarker.
marker.Lane
Lane1–20,
1–20,represents
representsAMPs
AMPs
1–20 SLOPE products.
1–20 SLOPE products.
These PCR products were purified and cloned into the pET23a-HH3C vector, result-
ing in pET23a-HH3C-AP(1-N). The recombinant plasmids were confirmed and trans-
formed into E. coli BL21 cells, resulting in EAP(1-N) strains (Figure 2).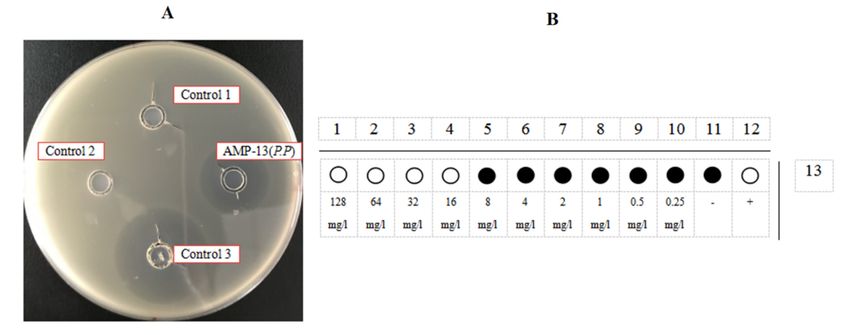
Microorganisms 2021, 9, 1858 5 of 15
Table 1. Sequences of AMPs.
Serial Number Amino Acid Sequence (MW: kDa) Nucleotide Sequence
ATTACGCCAGCAACACCCTTCACCCCCGCAATCATCACAGAGATCACGGCGGCGGT
AP00027(AMP1) ITPATPFTPAIITEITAAVIA(2.11)
CATAGCA
AP00150(AMP2) ILPWKWPWWPWRR(1.91) ATTTTACCTTGGAAGTGGCCATGGTGGCCGTGGCGTAGA
CGGGGCCTTCGTCGTCTGGGCCGTAAGATAGCGCACGGAGTAAAGAAATATGGACC
AP00155(AMP3) RGLRRLGRKIAHGVKKYGPTVLRIIRIAG(3.26)
CACCGTACTTAGAATAATTCGTATAGCCGGA
GGGTGGGGGAGTTTCTTCAAAAAGGCGGCTCATGTCGGCAAACACGTAGGTAAGGC
AP00166(AMP4) GWGSFFKKAAHVGKHVGKAALTHYL(2.71)
TGCTCTGACGCATTACTTG
GCTTGTTATTGCAGAATCCCCGCCTGCATTGCTGGAGAGCGTCGCTACGGGACCTGT
AP00176(AMP5) ACYCRIPACIAGERRYGTCIYQGRLWAFCC(3.45)
ATATATCAAGGCCGTTTGTGGGCCTTTTGTTGC
GTATTCCAATTTCTGGGAAAAATAATCCATCATGTAGGGAACTTCGTGCATGGATTTA
AP00276(AMP6) VFQFLGKIIHHVGNFVHGFSHVF(2.67)
GCCATGTTTTT
GGTTTGTTGCGGAAGGGAGGAGAGAAGATAGGTGAAAAACTGAAGAAAATAGGCC
AP00281(AMP7) GLLRKGGEKIGEKLKKIGQKIKNFFQKLVPQPEQ(3.88)
AGAAGATTAAGAACTTCTTTCAAAAGTTAGTACCGCAACCTGAGCAA
GGAATCATCAACACATTACAAAAATACTACTGCCGCGTCAGAGGGGGTCGTTGTGCT
AP00283(AMP8) GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK(5.16) GTTTTGAGTTGTCTGCCCAAGGAGGAACAGATAGGAAAATGTTCAACTAGAGGGCGT
AAATGCTGTCGCAGAAAGAAA
GGACTGCTGTGTTATTGCCGGAAAGGTCACTGCAAGCGTGGCGAGAGAGTACGGGG
AP00285(AMP9) GLLCYCRKGHCKRGERVRGTCGIRFLYCCPRR(3.76)
AACCTGTGGGATTCGCTTTTTATACTGTTGTCCCAGACGC
AP00351(AMP10) GLFDVIKKVASVIGGL(1.62) GGCCTGTTCGACGTGATTAAGAAGGTTGCTTCTGTGATCGGGGGACTT
GGGCGGTTCAAACGGTTTAGAAAGAAATTCAAAAAATTATTCAAGAAACTGAGCCC
AP00366(AMP11) GRFKRFRKKFKKLFKKLSPVIPLLHLG(3.28)
CGTTATTCCGTTGCTTCATCTGGGA
GGCGGATTAAGATCGCTGGGCCGGAAAATCCTTCGTGCCTGGAAGAAGTATGGCCC
AP00367(AMP12) GGLRSLGRKILRAWKKYGPIIVPIIRIG(3.13)
CATAATAGTACCTATCATACGGATTGGG
AP00445(AMP13) GFCRCLCRRGVCRCICTR(2.11) GGCTTTTGCAGATGTTTGTGTCGCAGAGGTGTCTGCCGTTGTATTTGCACAAGA
TTTTTTCATCATATTTTCAGAGGCATCGTTCATGTGGGCAAGACTATCCATCGGTTGG
AP00473(AMP14) FFHHIFRGIVHVGKTIHRLVTG(2.57)
TTACAGGC
GGTCTTGTGCGCAAAGGAGGTGAGAAGTTTGGCGAAAAGTTACGCAAAATCGGGC
AP00498(AMP15) GLVRKGGEKFGEKLRKIGQKIKEFFQKLALEIEQ(3.95)
AAAAAATCAAGGAGTTTTTTCAAAAATTAGCTCTGGAGATTGAACAA
GACTCCCATGCTAAGAGACACCACGGCTATAAACGTAAGTTTCACGAGAAGCACCA
AP00505(AMP16) DSHAKRHHGYKRKFHEKHHSHRGY(3.04)
TTCTCACCGTGGATAT
AP00513(AMP17) FLGGLIKIVPAMICAVTKKC(2.11) TTTCTGGGGGGCCTTATCAAGATCGTACCAGCCATGATATGCGCCGTCACGAAGAA ATGT
ATATGGTTGACGGCTCTTAAATTTCTGGGAAAACACGCTGCGAAGCACCTTGCGAAA
AP00516(AMP18) IWLTALKFLGKHAAKHLAKQQLSKL(2.85)
CAGCAGCTTAGCAAACTT
CAATGGGGAAGAAGATGCTGCGGCTGGGGTCCTGGACGCAGATACTGCCGCCGTTG
AP00519(AMP19) QWGRRCCGWGPGRRYCRRWC(2.54)
GTGT
AP00538(AMP20) WLNALLHHGLNCAKGVLA(1.93) TGGCTGAATGCCTTATTACACCACGGTCTGAACTGTGCCAAAGGTGTACTGGCC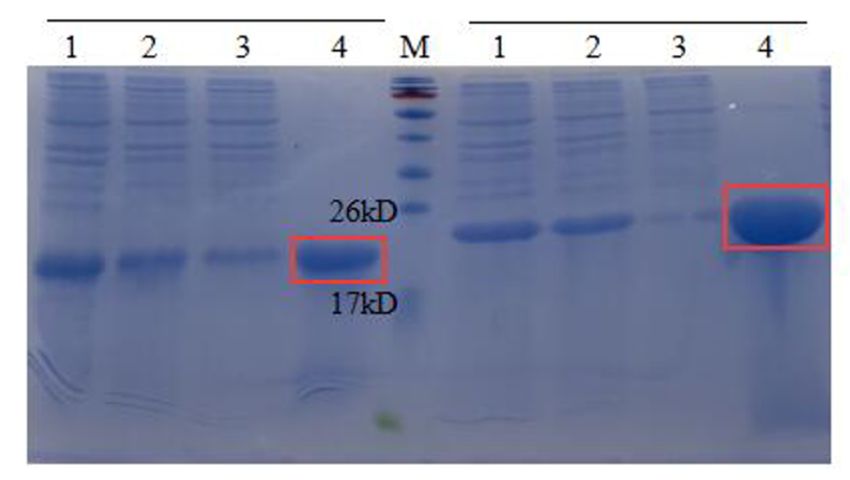
Microorganisms 2021, 9, 1858 6 of 15
These PCR products were purified and cloned into the pET23a-HH3C vector, resulting
Microorganisms 2021, 9, x FOR PEER REVIEW 7 of 16
in pET23a-HH3C-AP(1-N). The recombinant plasmids were confirmed and transformed
into E. coli BL21 cells, resulting in EAP(1-N) strains (Figure 2).
Figure 2. The construction of the AMP expression plasmids.
Figure 2. The construction of the AMP expression plasmids.
3.2. Expression of AMPs in E. coli and Purification
3.2. Expression of AMPs in EAP(1-N)
These recombinant E. coli and strains
Purification
were cultured in LB and induced with 0.5 mM
IPTG These ◦ C. Expression
at 16recombinant EAP(1-N)
of AMPs strains
was were cultured
assessed in LB and (Figure
by SDS-PAGE induced3). with
The0.5results
mM
IPTG at 16 °C. Expression of AMPs was assessed by SDS-PAGE (Figure
showed that AMPs (1, 2, 6, 13 and 16) were expressed at high levels in E. coli BL21 (DE3).3). The results
showed that AMPs (1,
These supernatants 2, 6,incubated
were 13 and 16) inwere expressed
a water bath at 80 ◦ C for
at high levels in E.
30 min, coli BL21 (DE3).
centrifuged at high
These
speed,supernatants
filtered throughwerea incubated in aand
0.45 µm filter, water bath at to
subjected 80Ni-NTA
°C for 30affinity
min, centrifuged at highIt
chromatography.
speed,
showed filtered through
that, after a 0.45temperature
the high μm filter, and subjected
treatment, to Ni-NTA affinity
heteroproteins in the chromatography.
supernatant were
Iteffectively
showed that, afterwhile
reduced the high temperature
target proteins were treatment, heteroproteins
unaffected. in the supernatant
The results showed that AMP13
and effectively
were AMP16 saturated
reducedthe column
while target(Figure
proteins4),were
and the amount The
unaffected. of visible
resultsheteroproteins
showed that
was low;
AMP13 and hence,
AMP16 these target proteins
saturated the column were deemed
(Figure suitable
4), and for subsequent
the amount of visiblestudies.
heteropro-The
fourth
teins waslanelow;inhence,
Figurethese
4 shows
targetthat the target
proteins were fusion
deemed protein bound
suitable successfully
for subsequent to the
studies.
nickel
The column.
fourth lane in Figure 4 shows that the target fusion protein bound successfully to the
nickel column.
Microorganisms 2021, 9, x FOR PEER REVIEW 8 of 16
Microorganisms 2021, 9, x1858
FOR PEER REVIEW 8 7of
of 16
15
Figure
Figure 3. 3. SDS-PAGE
SDS-PAGE (12%) (12%) analysis
analysis of of expression
expression of
of AMPs
AMPs ininE.E.coli.
coli.(A,B),
(A,B),lane lane1–18,
1–18,the
thesupernatants
supernatantsofofEAPEAP(1,
(1,2,2,6,6,7,7,8,
Figure
9, 10, 11, 12, 15, 16, 17, 18); lane 1–18*, the supernatant of EAP (1, 2, 6, 7, 8, 9, 10, 11, 12, 15, 16, 17, 18) heated to(1,
8, 9, 10, 3.
11,SDS-PAGE
12, 15, 16, (12%)
17, 18);analysis
lane of
1–18*, expression
the of AMPs
supernatant of in
EAP E. coli.
(1, 2, (A,B),
6, 7, 8, lane
9, 10,1–18,
11, the
12, supernatants
15, 16, 17, 18) of EAP
heated to 80 2,
80 ◦ C6,for
°C 7,
for
8, 9,
30 10, (C),
min; 11, 12, 15,1,16,
lane 3, 17,5,18);
4, 13, lane
14, 1–18*,
19, 20, thesupernatant
the supernatantofofEAP
EAP(1,(1,3,2,4,6,5,7,13,
8, 9,
14,10, 11,
19, 12,
20) 15, 16,to17,
heated 80 18)
°C
◦ heated
for 30 to 80 °C for
min.
30 min; (C), lane 1, 3, 4, 5, 13, 14, 19, 20, the supernatant of EAP (1, 3, 4, 5, 13, 14, 19, 20) heated to 80 C for 30 min.
30 min; (C), lane 1, 3, 4, 5, 13, 14, 19, 20, the supernatant of EAP (1, 3, 4, 5, 13, 14, 19, 20) heated to 80 °C for 30 min.
Figure 4. SDS-PAGE (12%) analysis of AMP (13, 16) purification. M, protein molecular weight marker. Left, lane 1, the
Figure 4. SDS-PAGE (12%) analysis of AMP (13, 16) purification. M, protein molecular weight marker. Left, lane 1, the
supernatant ofEAP
EAP13;13; laneanalysis
2, the
the supernatant
supernatant ofEAP
EAP 13heated
heatedtoto ◦ for 30 min; lane 3, the flow-through of EAP 13
80protein
Figure 4. SDS-PAGE
supernatant of (12%)
lane 2, of AMP (13, of 16) purification.
13 M,80 °CCfor molecular
30 min; laneweight marker. Left, lane
3, the flow-through of EAP1, the
13
with Ni2+-affinity;
Ni2+-affinity;
supernatant
with lane
of EAPlane 4,4, the
13; lane the AMP
2,AMP 13 purified
purifiedof
the supernatant
13 with Ni2+-affinity.
EAPNi2+-affinity.
with Right,
13 heated to Right,
80 °C forlane
lane 301,1,
min;the supernatant
the supernatant
lane of EAP
EAP 16;
3, the flow-through
of 16; lane
lane
of EAP2, the
2, the
13
supernatant
with of EAP lane
Ni2+-affinity;
supernatant 16 heated
4, theto
heated AMP
to 80 ◦°C
80 C13for 30
30 min;
purified
for lane
min;with 3,3,the
theflow-through
laneNi2+-affinity. Right,oflane
flow-through ofEAP
EAP1,16the
16with Ni2+-affinity;
supernatant
with lane
of EAP
Ni2+-affinity; 4, 4,
16;
lane the
laneAMP
the 16
2, the
AMP
supernatant
16 purified of
withEAP 16 heated
Ni2+-affinity.
purified with Ni2+-affinity. to 80 °C for 30 min; lane 3, the flow-through of EAP 16 with Ni2+-affinity; lane 4, the AMP
16 purified with Ni2+-affinity.
Afterfusion
After fusionproteins
proteinswere
werepurified
purifiedbybyNi-NTA
Ni-NTAaffinity
affinity chromatography,
chromatography, they they were
were
cleaved
After by HRV
fusion 3C to remove
proteins were the CL7
purified protein.
by The
Ni-NTA collected
affinity components
chromatography,
cleaved by HRV 3C to remove the CL7 protein. The collected components were analysed were analysed
they were
byTricine-SDS-PAGE
by Tricine-SDS-PAGE
cleaved (Figure 5),
by HRV 3C to (Figure
remove 5), and
the and the
CL7the results
protein.
results ofcollected
The
of restriction
restriction digestionwere
components
digestion were observed.
wereobserved.
analysed
Lanes
by 3,3,44and
and55show
Tricine-SDS-PAGE
Lanes showthe thedigestion
digestion
(Figure 5), andsupernatant, HRV
the resultsHRV
supernatant, of 3CCleavage
Cleavage
restriction
3C buffer
digestion
buffer and1M
were
and 1MNaCl,
NaCl,
observed.
respectively.
Lanes 3, 4 and
respectively. The HRV 3C
5 show
The HRV 3C protease
the protease band
digestionband can be
be observed
supernatant,
can observed
HRV 3C along with the
Cleavage
along with the target
buffer AMPs.
andAMPs.
target The
1M NaCl,
The
results showed
respectively. Thethat
HRVthe3Csize of the fusion
protease protein
band can was altered
be observed alongsignificantly before
with the target and after
AMPs. The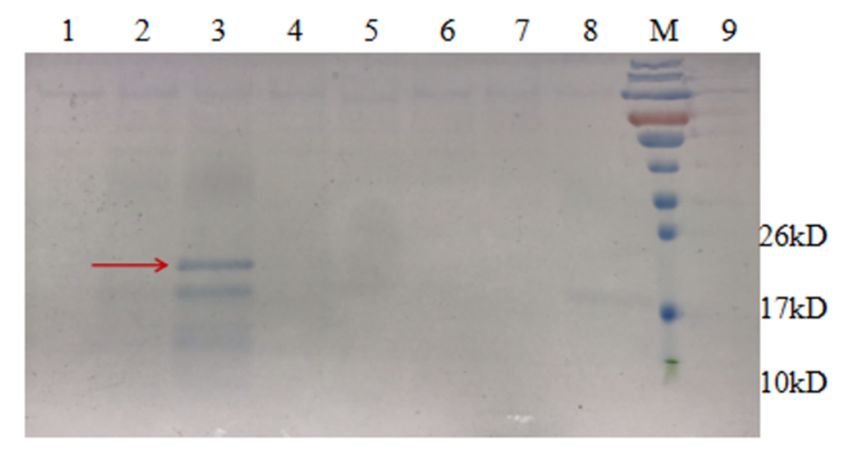
Microorganisms 2021, 9, x FOR PEER REVIEW 9 of 16
Microorganisms 2021, 9, 1858 8 of 15
results showed that the size of the fusion protein was altered significantly before and after
digestion.
digestion.TheThelarger
largerbands
bandsininthe
thedigestion
digestionsupernatant
supernatantandandthe
theeluate
eluateafter
afterdigestion
digestion
correspond
correspond to the HRV 3C protein which is ~22 kDa in size. The band markedby
to the HRV 3C protein which is ~22 kDa in size. The band marked bythe
thebox
box
ininFigure 5 is the target AMP band. After digestion, only a small part of the target
Figure 5 is the target AMP band. After digestion, only a small part of the target AMPs AMPs
was
waspresent
presentininthe
thedigestion
digestionsupernatant.
supernatant.
Figure 5. Tricine SDS-PAGE analysis of AMPs cleaved by HRV 3C. M, protein molecular weight
Figure 5. Tricine
marker. Lane 1,SDS-PAGE analysisbefore
the nickel column of AMPs cleavedlane
digestion; by HRV
2, the 3C. M, column
nickel protein after
molecular weight
digestion and
marker. Lane 1, the nickel column before digestion; lane 2, the nickel column after digestion and
elution; lane 3, the digestion supernatant; lane 4, HRV 3C Cleavage buffer; lane 5, 1M NaCl.
elution; lane 3, the digestion supernatant; lane 4, HRV 3C Cleavage buffer; lane 5, 1M NaCl.
Theoretically, target AMPs should be in the digestion supernatant. However, it was
foundTheoretically, target
that part of the AMPsAMPs should
could not bebecompletely
in the digestion
releasedsupernatant. However,
into the digestion it was
supernatant
found that part of the AMPs could not be completely released into the
after digestion. It remained bound to the nickel column. Hence, 1 M NaCl was used to digestion superna-
tant afterthe
release digestion. It remained
target AMPs as much bound to the nickel
as possible. column.protein
The collected Hence,sample
1 M NaClwaswas used
incubated
toinrelease the target AMPs◦ as much as possible. The collected protein
a water bath at 80 C for 30 min to remove heteroproteins and other contaminants sample was incu-
bated in a water
introduced withbath
theatHRV
80 °C3Cforprotease.
30 min to remove heteroproteins
Supernatants and other
were collected contaminants
after temperature
introduced with the HRV 3C protease. Supernatants were collected
treatment, concentrated, and desalted to obtain target AMPs. The protein concentration after temperature
treatment,
measuredconcentrated,
by the Bradford andmethod
desalted
was to 0.2
obtain target0.3
mg/mL, AMPs.
mg/mL, The0.2
protein
mg/mL, concentration
0.6 mg/ML
measured by the Bradford method was 0.2 mg/mL, 0.3 mg/mL,
and 0.7 mg/mL for AMP1, AMP 2, AMP 6, AMP 13 and AMP 16, respectively. 0.2 mg/mL, 0.6 mg/Ml and
0.7 mg/mL for AMP1,
Judging from the AMP 2, AMP 6,results
purification AMP (Figure
13 and AMP
6), the16, respectively.
purity of the obtained proteins
wasJudging
good, andfrom the were
there purification
no otherresults (Figure 6), The
heteroproteins. the purity
bands of the obtained to
corresponding proteins
the five
Microorganisms 2021, 9, x FOR PEER REVIEW 10 of 16
was good,
target and there
proteins werewere no other
all larger thanheteroproteins.
the theoreticalThe bands
value, corresponding
consistent with the to results
the fiveof
target
Tricine proteins
SDS-PAGE,wereandall larger than that
suggesting the theoretical value,
target proteins may consistent with the results of
form multimers.
Tricine SDS-PAGE, and suggesting that target proteins may form multimers.
Figure
Figure 6. Tricine
6. Tricine SDS-PAGE
SDS-PAGE analysis
analysis of AMPofpurification.
AMP purification. Lane
Lane 1-5, the 1-5, the
purified purified
AMP AMP
1, 2, 6, 13 1, 2, 6, 13
and 16.
and 16.
3.3. AMP Activity Testing
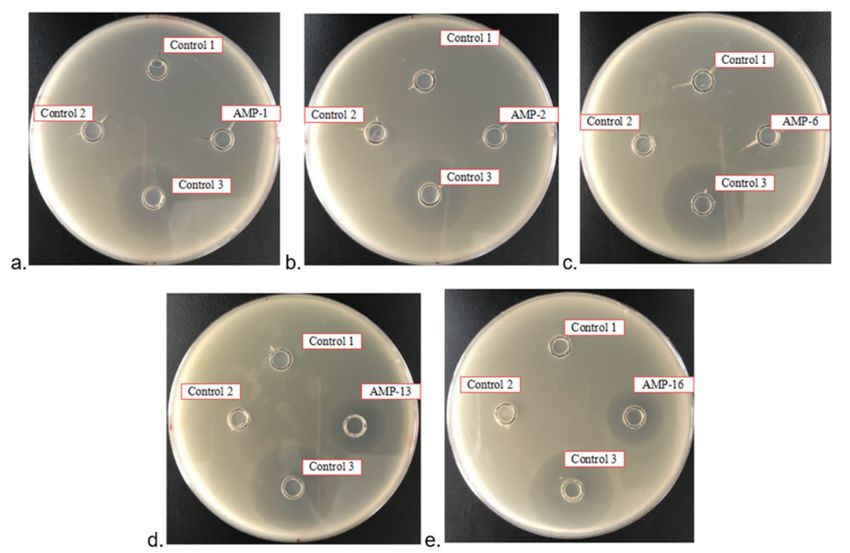
There were no inhibition zones around negative controls 1 and 2, but there was an
inhibition zone around positive control 3 (Figure 7). There were no inhibition zones
around test samples AMP1, AMP 2 and AMP 6, but there was an inhibition zone around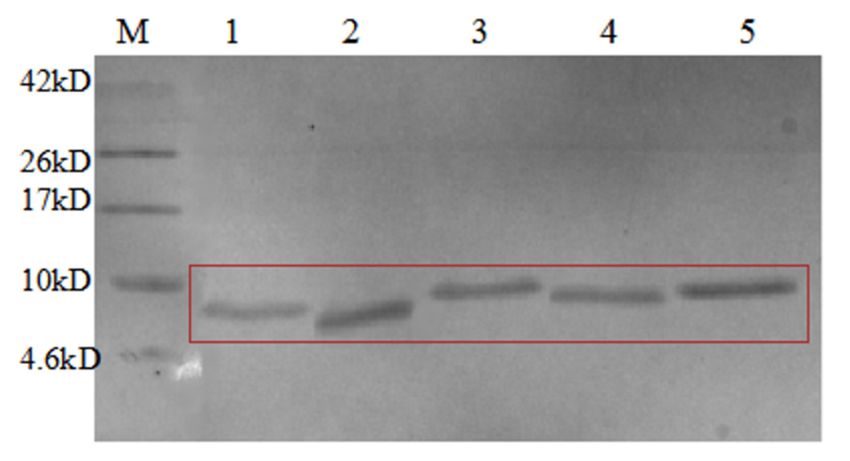
Microorganisms 2021, 9, 1858 9 of 15
Figure 6. Tricine SDS-PAGE analysis of AMP purification. Lane 1-5, the purified AMP 1, 2, 6, 13 and 16.
3.3. AMP Activity Testing
3.3. AMP Activity Testing
There were
There were no
no inhibition
inhibition zones
zones around
around negative
negative controls
controls 11 and
and 2,
2, but
but there
there was
was an
an
inhibition zone around positive control 3 (Figure 7). There were no inhibition zones
inhibition zone around positive control 3 (Figure 7). There were no inhibition zones around
around
test test samples
samples AMP1,
AMP1, AMP AMP
2 and 2 and
AMP AMP
6, but 6, but
there wasthere was an inhibition
an inhibition zone aroundzonesamples
around
samples AMP
AMP 13 and 16. 13 and 16.
Figure 7.
Figure 7. Inhibition zone tests of AMP 1, 2, 6, 13 and 16. Control 1, HRV 3C Cleavage Buffer; Control 2, HRV 3C Cleavage
Buffer with
Buffer with 11 M
M NaCl;
NaCl; Control
Control 3,
3,Positive
Positivecontrol,
control,ampicillin
ampicillinantibiotic
antibioticat
ataaconcentration
concentrationofof200
200mg/L.
mg/L. (a) Inhibition zone
zone
tests of
tests of AMP1;
AMP1; (b)(b) Inhibition
Inhibitionzone
zonetests
testsofofAMP2;
AMP2;(c)(c)Inhibition
Inhibitionzone
zone tests ofof
tests AMP6; (d)(d)
AMP6; Inhibition zone
Inhibition tests
zone of AMP13;
tests (e)
of AMP13;
Inhibition
(e) zone
Inhibition tests
zone of AMP16.
tests of AMP16.Every oxford
Every cupcup
oxford contains 100100
contains μLµLliquid.
liquid.
The MIC results (Figure 8) showed that the minimum increase in OD600 value for
AMP1 was 128 mg/L, and the minimum increase in OD600 for AMP2 and AMP6 was
>128 mg/L. This means that AMP1, AMP2 and AMP6 had no inhibition activity against
testing the microorganisms. The minimum concentration of purified AMP13 and AMP16
that inhibited an increase in the OD600 of the bacterial solution was 32 mg/L and 64 mg/L,
respectively. The MIC of AMP16 was 24.2 µM.
3.4. Expression of AMPs in P. pastoris and Purification
The P. pastoris expression plasmids pHBM905BDM-HH3C and pPICZα-HH3C were
constructed similarly to the E. coli expression vector pET23a-HH3C. Fragments of AMP1,
AMP2, AMP6, AMP13 and AMP16 were cloned into pHBM905BDM-HH3C and pPICZα-
HH3C, resulting in ten recombinant plasmids. They were transformed into P. pastoris,
resulting in corresponding recombinant strains PBEAP(N) and PZEAP(N), respectively.
These strains were cultured and induced. The results of shake flask experiments showed
that the expression supernatants of PBEAP (1, 2, 6, 13, 16) did not produce the expected
AMPs. However, the results of PZEAP (1, 2, 6, 13, 16) experiments revealed a band of the
expected product from PZEAP13 (Figure 9).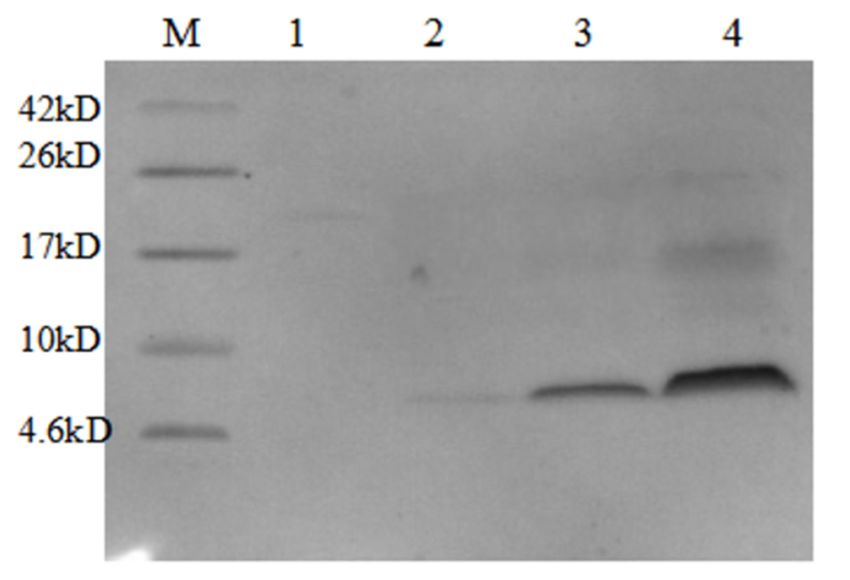
The MIC results (Figure 8) showed that the minimum increase in OD600 value for
AMP1 was 128 mg/L, and the minimum increase in OD600 for AMP2 and AMP6 was >128
mg/L. This means that AMP1, AMP2 and AMP6 had no inhibition activity against testing
the microorganisms. The minimum concentration of purified AMP13 and AMP16 that in-
Microorganisms 2021, 9, 1858 10 of 15
hibited an increase in the OD600 of the bacterial solution was 32 mg/L and 64 mg/L, respec-
tively. The MIC of AMP16 was 24.2 μM.
Figure 8. The schematic diagram of a 96-well plate measuring MIC (data taken from three parallel
experiments). Horizontal direction is AMP 1, 2, 6, 13, 16 test rows; Vertical direction 1–10 is the final
concentration of 128 mg/L to 0.25 mg/L of these AMPs, respectively; 11 is the negative control; 12 is
a positive control, the 20 mg/L ampicillin.
3.4. Expression of AMPs in P. pastoris and Purification
The P. pastoris expression plasmids pHBM905BDM-HH3C and pPICZα-HH3C were
constructed similarly to the E. coli expression vector pET23a-HH3C. Fragments of AMP1,
AMP2, AMP6, AMP13 and AMP16 were cloned into pHBM905BDM-HH3C and pPICZα-
HH3C, resulting in ten recombinant plasmids. They were transformed into P. pastoris, re-
sulting in corresponding recombinant strains PBEAP(N) and PZEAP(N), respectively.
These strains were cultured
Theschematic
schematic diagramandof induced. The results of shake flask experiments showed
Figure
Figure 8.8.The diagram of aa96-well
96-wellplate
plate measuring
measuring MIC(data
MIC (datataken
takenfrom
fromthree
three parallel
parallel
that the expression
experiments).Horizontal supernatants
Horizontaldirection of
directionisisAMPPBEAP (1, 2,
AMP1,1,2,2,6,6,13, 6, 13,
13,1616test 16)
testrows;did not
rows;Vertical produce
Verticaldirection the
direction1–10 expected
1–10isisthe
thefinal
final
experiments).
AMPs. However,
concentration
concentration the
ofof128
128 results
mg/L
mg/L of mg/L
toto0.25
0.25PZEAP
mg/L ofof(1, 2,AMPs,
these
these 6,AMPs,
13, 16) experiments
1111isisrevealed
respectively;
respectively; a band
thenegative
the negative of 12
control;
control; the
12isis
expected
a apositive
positive product
control, from
control,the PZEAP13
the2020mg/L
mg/L (Figure 9).
ampicillin.
ampicillin.
3.4. Expression of AMPs in P. pastoris and Purification
The P. pastoris expression plasmids pHBM905BDM-HH3C and pPICZα-HH3C were
constructed similarly to the E. coli expression vector pET23a-HH3C. Fragments of AMP1,
AMP2, AMP6, AMP13 and AMP16 were cloned into pHBM905BDM-HH3C and pPICZα-
HH3C, resulting in ten recombinant plasmids. They were transformed into P. pastoris, re-
sulting in corresponding recombinant strains PBEAP(N) and PZEAP(N), respectively.
These strains were cultured and induced. The results of shake flask experiments showed
that the expression supernatants of PBEAP (1, 2, 6, 13, 16) did not produce the expected
AMPs. However, the results of PZEAP (1, 2, 6, 13, 16) experiments revealed a band of the
expected product from PZEAP13 (Figure 9).
Figure 9. SDS-PAGE (12%) analysis of expression of AMP13 in Pichia pastoris. M, protein molecular
weight marker. Lane 3, AMP produced by P. pastoris.
The supernatant of strain PZEAP13 was subsequently used for purification, and
the results were assessed by SDS-PAGE (Figure 10) and Tricine SDS-PAGE (Figure 11).
Comparative analysis before and after temperature treatment using SDS-PAGE (Figure 10,
lanes 1 and 2) showed that high temperature treatment did not denature or decrease
the amount of the target fusion protein; the change in the size of the protein in lanes
3 and 4 clearly show that AMPs were successfully released by HRV 3C protease. The
specific cleavage site was cut (lanes 5 and 6), and there was a band corresponding to HRV
3C protease.results were assessed by SDS-PAGE (Figure 10) and Tricine SDS-PAGE (Figure 11). Com-
parative
parative analysis
analysis before
before and
and after
after temperature
temperature treatment
treatment using
using SDS-PAGE
SDS-PAGE (Figure
(Figure 10,10,
lanes
lanes 1 and 2) showed that high temperature treatment did not denature or decrease the
1 and 2) showed that high temperature treatment did not denature or decrease the
amount
amount ofof the
the target
target fusion
fusion protein;
protein; the
the change
change in
in the
the size
size of
of the
the protein
protein in
in lanes
lanes 33 and
and 44
clearly show that AMPs were successfully released by HRV 3C protease.
clearly show that AMPs were successfully released by HRV 3C protease. The specific The specific
Microorganisms 2021, 9, 1858 cleavage
cleavage site
site was
was cut
cut (lanes
(lanes 55 and
and 6),
6), and
and there
there was
was aa band
band corresponding
corresponding to to HRV
HRV11 of3C15
3C
protease.
protease.
SDS-PAGE (12%)
Figure 10. SDS-PAGE (12%) analysis
analysisof ofHRV
HRV3C 3Cprotease
proteasecleavage
cleavageofofAMP13.
AMP13.M, M,protein
proteinmolecular
molecularweight
weightmarker.
marker. Lane 1,
Lane
Figure 10. SDS-PAGE (12%) analysis of HRV 3C protease cleavage of AMP13. M, protein ◦ to
molecular weight marker. Lane
1,
thethe supernatant
supernatant fromfrom
P. P. pastoris;
pastoris; lane lane
2, 2,supernatant
the the supernatant
from from
P. P. pastoris
pastoris heatedheated
to 80 C 80 30
for °C min;
for 30 min;
lane 3, lanenickel
the 3, thecolumn
nickel
1, the supernatant from P. pastoris; lane 2, the supernatant from P. pastoris heated to 80 °C for 30 min; lane 3, the nickel
column
before before digestion; lane 4, the nickel after
column after digestion and lane
elution; lane 5, the digestion supernatant; lane3C6,
columndigestion; lane 4, the
before digestion; nickel
lane column
4, the nickel columndigestion and elution;
after digestion 5, the
and elution; digestion
lane supernatant;
5, the digestion lane 6, HRV
supernatant; lane 6,
HRV 3C Cleavage
Cleavage wash wash buffer.
buffer.
HRV 3C Cleavage wash buffer.
Figure 11. Tricine SDS-PAGE analysis of HRV 3C protease cleavage AMP13. M, protein molecular weight marker. Lane 1,
the supernatant from P. pastoris; lane 2, the supernatant from P. pastoris heated to 80 ◦ C for 30 min; lane 3, the nickel column
before digestion; lane 4, the nickel column after digestion and elution; lane 5, the digestion supernatant; lane 6–9, HRV 3C
cleavage wash buffer with NaCl.
Similarly, Tricine-SDS-PAGE analysis showed that high temperature treatment did not
denature or decrease the amount of the target fusion protein (Figure 11, lanes 1 and 2). The
change in the size of the protein in lanes 3 and 4 clearly shows that AMPs were successfully
recovered by HRV 3C protease. The specific cleavage site was cut, and bands marked by
boxes in lanes 5 to 9 correspond to the target AMPs released following cleavage. Figure 12
also confirmed these results.Similarly, Tricine-SDS-PAGE analysis showed that high temperature treatment did
not denature or decrease the amount of the target fusion protein (Figure 11, lanes 1 and
2). The change in the size of the protein in lanes 3 and 4 clearly shows that AMPs were
successfully recovered by HRV 3C protease. The specific cleavage site was cut, and bands
Microorganisms 2021, 9, 1858 12 of 15
marked by boxes in lanes 5 to 9 correspond to the target AMPs released following cleavage.
Figure 12 also confirmed these results.
Figure 12. Tricine SDS-PAGE analysis of purification of AMP 13. M, protein molecular weight marker.
Figure 12. Tricine SDS-PAGE analysis of purification of AMP 13. M, protein molecular weight
Lane 1–4,
marker. the1–4,
Lane purified AMP13AMP13
the purified produced by P. pastoris.
produced by P. pastoris.
The E. coli DH5α mixed bacteria plate was prepared as described above. Two negative
The E. coli DH5α mixed bacteria plate was prepared as described above. Two nega-
controls (HRV 3C Cleavage Buffer and 1 M NaCl) and a positive control (2 mg/mL ampi-
tive controls (HRV 3C Cleavage Buffer and 1 M NaCl) and a positive control (2 mg/mL
cillin) were included. According to the growth of the mixed bacteria plate after culture,
ampicillin) were included. According to the growth of the mixed bacteria plate after cul-
there were no inhibition zones around negative controls 1 and 2, but there was an inhibition
ture, there were no inhibition zones around negative controls 1 and 2, but there was an
zone around positive control 3. Apparently, AMP13 showed inhibition activity. Using
inhibition zone around positive control 3. Apparently, AMP13 showed inhibition activity.
the MIC measurement method described above, the minimum concentration of purified
Using the MIC measurement method described above, the minimum concentration of pu-
AMP13 produced by PZEAP13 that inhibited an increase in the OD600 of the bacterial
rified AMP13 produced by PZEAP13 that inhibited an increase in the OD600 of the bacterial
solution was 16 mg/L, equating to an MIC for E. coli of 8.1 µM.
solution was13B
Figure 16 mg/L, equatingdiagram
is a schematic to an MIC for 96-well
of the E. coli ofplate
8.1 μM.
MIC experiment (data are from
Figure 13B is a schematic diagram of the 96-well plate
three parallel replicates). The horizontal direction is AMP13 from MIC experiment (data
P. pastoris, theare from
vertical
three parallel
direction replicates).
is the The horizontal
final peptide direction
concentration is AMP13
(128 mg/L from
to 0.25 P. pastoris,
mg/L), the negative
11 is the vertical
direction is the final peptide concentration (128 mg/L to 0.25 mg/L), 11 is the
control and 12 is the positive control (20 mg/L ampicillin). The results showed that the negative
control
minimum andconcentration
12 is the positive control
of AMP13 (20 P.
from mg/L ampicillin).
pastoris The results
that inhibited showed
an increase in thethat
OD the
600
minimum concentration of AMP13
of the bacterial solution was 16 mg/L. from P. pastoris that inhibited an increase in the OD 600
of the bacterial solution was 16 mg/L.Microorganisms 2021,
Microorganisms 9, x1858
2021, 9, FOR PEER REVIEW 1413ofof16
15
Figure 13. The activity test of AMP13 produced by P. pastoris. (A). Inhibition zone tests of AMP13. Control 1, HRV 3C
Figure 13. The activity test of AMP13 produced by P. pastoris. (A). Inhibition zone tests of AMP13. Control 1, HRV 3C
CleavageBuffer;
Cleavage Buffer;Control
Control2,2,HRV
HRV3C 3CCleavage
CleavageBuffer
Bufferwith
with1 1MMNaCl;
NaCl;Control
Control3,3,Positive
Positive control,
control, ampicillin
ampicillin antibiotic
antibiotic at at
a
a concentration of 200 mg/L. Every oxford cup contains 100 µL liquid. (B). The schematic diagram of
concentration of 200 mg/L. Every oxford cup contains 100 μL liquid. (B). The schematic diagram of a 96-well plate meas- a 96-well plate
measuring
uring MIC taken
MIC (data (data taken from parallel
from three three parallel experiments).
experiments). Horizontal
Horizontal direction
direction is AMP is 13
AMPtest13 test rows;
rows; Vertical
Vertical direction
direction 1–10
is the is
1–10 final
theconcentration of 128 of
final concentration mg/L
128 to 0.25 to
mg/L mg/L
0.25ofmg/L
these of
AMPs,
theserespectively; 11 is the 11
AMPs, respectively; negative control; 12
is the negative is a positive
control; 12 is a
control,
positivethe 20 mg/L
control, the ampicillin.
20 mg/L ampicillin.
4. Discussion
4. Discussion
The AMP sequences chosen in this experiment were selected from an AMP library,
cloned,The AMP sequences
expressed, chosen using
and screened in thisaexperiment
workflow that wereisselected from
universal. AMP an AMP
geneslibrary,
were
cloned, expressed, and screened using a workflow
cloned into pET-23a, pPICZα and pHBM905BDM vectors, and expressed and that is universal. AMP genes were
screened
cloned into pET-23a, pPICZα and pHBM905BDM vectors, and
using E. coli and P. pastoris systems. The purification process included high-temperatureexpressed and screened
using E. coli
treatment of and P. pastoris
the protein systems. The
supernatant, HRVpurification
3C protease process
enzyme included high-temperature
digestion on the nickel
treatment of the protein supernatant, HRV 3C protease enzyme
column, subsequent temperature treatment, protein concentration and desalination. digestion on the nickel
This
column, subsequent temperature treatment, protein concentration and desalination. This
approach has multiple advantages: (1) The SLOPE method is superior to other methods.
approach has multiple advantages: (1) The SLOPE method is superior to other methods.
When obtaining AMP gene fragments using the SLOPE method, the high cost is greatly
When obtaining AMP gene fragments using the SLOPE method, the high cost is greatly
reduced compared with traditional gene synthesis, and it can generate gene fragments
reduced compared with traditional gene synthesis, and it can generate gene fragments
with sticky ends in one step. Following processing of the ready-to-use vector, the cloning
with sticky ends in one step. Following processing of the ready-to-use vector, the cloning
efficiency can also be improved because the large-scale screening of AMPs can be
efficiency can also be improved because the large-scale screening of AMPs can be achieved
achieved without PCR to obtain each target gene fragment. (2) Most of the AMPs and the
without PCR to obtain each target gene fragment. (2) Most of the AMPs and the CL7 fusion
CL7 fusion protein tag are resistant to high temperatures, which makes the purification
protein tag are resistant to high temperatures, which makes the purification process easier
process easier when using an E. coli expression system. After appropriate temperature
when using an E. coli expression system. After appropriate temperature treatment of the
treatment of the expression supernatant, most of the endogenous proteins are denatured
expression supernatant, most of the endogenous proteins are denatured and precipitated
and precipitated due to temperature intolerance, while the fusion protein can tolerate high
due to temperature intolerance, while the fusion protein can tolerate high temperature
temperature treatment.
treatment. After high-speedAftercentrifugation,
high-speed centrifugation, some heteroproteins
some heteroproteins are precipitatedare andprecipi-
can be
tated and can be separated. This affords a useful preliminary purification
separated. This affords a useful preliminary purification step, which makes the procedure step, which
makes
cheap,the procedure
highly efficient cheap, highly efficient
and convenient. andmethod
(3) The convenient. (3) The method
is compatible with bothis compatible
E. coli and
with both E. coli and P. pastoris expression systems. The prokaryotic
P. pastoris expression systems. The prokaryotic E. coli expression system has advantages, E. coli expression sys-
tem has advantages,
including a rapid growth including
cycle,a lowrapid growth and
pollution cycle, low pollution
simple and simple
culture conditions, cultureit
making
conditions, making it suitable for fast screening and expression of
suitable for fast screening and expression of AMPs. The eukaryotic P. pastoris expression AMPs. The eukaryotic
P. pastoris
system is expression
more suited system
to theissecretion
more suitedand to the secretion
expression and expression
heterologous heterologous
proteins, especially
proteins,
where the copy number of the target gene sequence is increased, expression conditions ex-
especially where the copy number of the target gene sequence is increased, are
pression
optimised conditions are optimised
and high-density and high-density
fermentation is employed. fermentation
These advantagesis employed.
make this These ad-
system
vantages
well suitedmake thisindustrial
to the system well suited toofthe
production industrial production of AMPs.
AMPs.
After
Afterconstruction
constructionof ofthe
theE. E.coli
coliexpression
expressionvector,
vector, itit was
was transformed
transformed into into the
the E.
E. coli
coli
BL21 (DE3) strain for expression in shake flasks. During this process,
BL21 (DE3) strain for expression in shake flasks. During this process, the number of the number of colo-
nies on each
colonies plateplate
on each waswas notnotsignificantly
significantly different, while
different, while thethemorphology
morphologyofofcolonies
colonieson on
plates containing samples 1, 2, 6, 7, 11, 13 and 16 did differ, and colonies
plates containing samples 1, 2, 6, 7, 11, 13 and 16 did differ, and colonies were significantly were significantlyMicroorganisms 2021, 9, 1858 14 of 15
smaller than for other samples. Bacterial culture took 8 h for the OD600 to reach 0.6 for
EAP1, 2, 6, 11 and 16, and 9 h for EAP7 and 13. After induction with 1 mM IPTG for 24 h
at 18 ◦ C, the number of bacteria collected was significantly less for these samples than for
others. Thus, we speculated that, although the fusion expression strategy was successfully
employed, the fusion protein expressed by the host had a certain inhibitory effect on host
cell growth. This phenomenon can be utilized to screen recombinant strains. If the amount
of transformed plasmid and the volume of transformed competent cells are the same, then
under the same culture conditions, smaller colonies on the transformed plate could express
AMPs at higher levels than those larger colonies. With sufficient data, this feature could be
used to initially select strains for expression, or to select colonies with better expression
potential on the same transformation plate. Similarly, when preparing bacterial biomass
in shake flasks, and all other conditions are optimised, shake flasks displaying a slower
increase in the OD600 value are more likely to express larger amounts of heterologous
fusion proteins. After the screening of highly expressed AMPs using E. coli, these AMP
genes were cloned into the P. pastoris expression vectors pPICZα and pHBM905BDM,
resulting in five constructs. The following experiments showed that only pPICZα- HH3C
-13 successfully expressed the target protein.
Activity tests indicated that two AMPs produced by E. coli could yield inhibition zones,
along with one AMP produced in the yeast expression system. In the MIC experiment,
samples not yielding inhibition zones displayed no activity, consistent with the published
literature. Importantly, the inhibition zone experiment requires high antibacterial activity.
The AMPs with effective MICs using the E. coli expression system were EAP13 and EAP16.
After purification of AMP13 from P. pastoris, we observed a clear circle of inhibition in the
antibacterial experiment, and the effective MIC was 16 mg/mL. AMP1, AMP2 and AMP6
obtained by expression and purification in E. coli did not exhibit detectable antibacterial
activity. We eliminated the possible influence of the expression conditions and purification
process on antibacterial peptidase activity. After HRV 3C protease digestion, there were
two amino acid residues (G and P) appended at the N-terminus of the AMPs that may
affect its activity. The MICs of the purified AMPs obtained in this study were lower than
those reported previously. AMP activity can be optimised by manipulating the expression
conditions and the purification protocol. The purity of the obtained AMPs can greatly
improve the MIC. The AMPs can also be characterized in depth, such as the effects of pH,
temperature and metal ions on enzyme activity, along with the activity of AMPs against
various Gram-negative and Gram-positive bacteria and fungi.
Author Contributions: Conceptualisation, all authors; experimental work, Y.Z., Z.L. and L.L.; super-
vision, Y.W. and L.M.; writing—original draft preparation, B.R.; review and editing, all authors. All
authors have read and agreed to the published version of the manuscript.
Funding: This research was funded by the China National Key Research and Development (R&D)
Program (2021YFC2100100), Open Funding Project of the State Key Laboratory of Biocatalysis and
Enzyme Engineering (SKLBEE2018003), project of Yunnan local undergraduate universities (part)
(202001BA070001-086).
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Data Availability Statement: All relevant data of this study are presented. Additional data can be
provided upon request.
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Braam, J.F.; van Dommelen, L.; Henquet, C.J.M.; van de Bovenkamp, J.H.B.; Kusters, J.G. Multidrug-resistant Mycoplasma
genitalium infections in Europe. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 1565–1567. [CrossRef] [PubMed]
2. Magana, M.; Muthuirulan, P.; Ana, L.S.; Leon, L.; Michael, F.; Anastasios, I.; Marc, A.G.; Yiorgos, A.; Steven, B.; Andrew, L.F.; et al.
The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [CrossRef]Microorganisms 2021, 9, 1858 15 of 15
3. Wenyi, L.; Frances, S.; Neil, M.S.; John, D.W. Chemically modified and conjugated antimicrobial peptides against superbugs.
Chem. Soc. Rev. 2021, 50, 4932–4973.
4. Harkins, C.P.; Pichon, B.; Doumith, M.; Parkhill, J.; Westh, H.; Tomasz, A.; de Lencastre, H.; Bently, S.D.; Kearns, A.M.; Holden,
M.T.G. Methicillin-resistant Staphylococcus aureus emerged long before the introduction of methicillin into clinical practice. Genome.
Biol. 2017, 18, 130. [CrossRef] [PubMed]
5. Ansari, A.; Zohra, R.R.; Tarar, O.M.; Oader, S.A.U.; Aman, A. Screening, purification and characterization of thermostable,
protease resistant Bacteriocin active against methicillin resistant Staphylococcus aureus (MRSA). BMC Microbiol. 2018, 18, 192.
[CrossRef] [PubMed]
6. Nuti, R.; Goud, N.S.; Saraswati, A.P.; Alvala, R.; Alvala, M. Antimicrobial Peptides: A Promising Therapeutic Strategy in Tackling
Antimicrobial Resistance. Curr. Med. Chem. 2017, 24, 4303–4314. [CrossRef] [PubMed]
7. Vassylyeva, M.N.; Klyuyev, S.; Vassyley, A.D.; Wesson, H.; Zhang, Z.; Renfrow, M.B.; Wang, H.B.; Higgins, N.P.; Chow, L.T.;
Vassylyev, D.G. Efficient, ultra-high-affinity chromatography in a one-step purification of complex proteins. Proc. Natl. Acad. Sci.
USA 2017, 114, E5138–E5147. [CrossRef] [PubMed]
8. Wibowo, D.; Zhao, C.X. Recent achievements and perspectives for large-scale recombinant production of antimicrobial peptides.
Appl. Microbiol. Biotechnol. 2019, 103, 659–671. [CrossRef] [PubMed]
9. Sang, M.; Wei, H.; Zhang, J.X.; Wei, Z.H.; Xu, X.L.; Chen, Y.; Zhu, G.Q. Expression and characterization of the antimicrobial
peptide ABP-dHC-cecropin A in the methylotrophic yeast Pichia pastoris. Protein Expr. Purif. 2017, 140, 44–51. [CrossRef]
[PubMed]
10. Park, S.I.; Kim, J.W.; Yoe, S.M. Purification and characterization of a novel antimicrobial peptide from black soldier fly (Hermetia
illucens) larvae. Dev. Comp. Immunol. 2015, 52, 98–106. [CrossRef] [PubMed]
11. Saint, J.K.; Henderson, K.D.; Chrom, C.L.; Abiuso, L.E.; Renn, L.M.; Caputo, G.A. Effects of Hydrophobic Amino Acid Substitu-
tions on Antimicrobial Peptide Behavior. Probiotics Antimicrob. Proteins 2018, 10, 408–419. [CrossRef] [PubMed]
12. Almaas, H.; Eriksen, E.; Sekse, C.; Comi, I.; Flengsrud, R.; Holm, H.; Jensen, E.; Jacobsen, M.; Langsrud, T.; Vegarud, G.E.
Antimicrobial peptides derived from caprine whey proteins, by digestion with human gastrointestinal juice. Br. J. Nutr. 2011, 106,
896–905. [CrossRef] [PubMed]
13. Wang, M.; Zheng, K.; Lin, J.L.; Huang, M.H.; Ma, Y.; Li, S.; Luo, X.C.; Wang, J.F. Rapid and efficient production of cecropin
A antimicrobial peptide in Escherichia coli by fusion with a self-aggregating protein. BMC Biotechnol. 2018, 18, 62. [CrossRef]
[PubMed]
14. Daly, R.; Hearn, M.T. Expression of heterologous proteins in Pichia pastoris: A useful experimental tool in protein engineering and
production. J. Mol. Recognit. 2005, 18, 119–138. [CrossRef] [PubMed]
15. Esposito, D.; Chatterjee, D.K. Enhancement of soluble protein expression through the use of fusion tags. Curr. Opin. Biotechnol.
2006, 17, 353–358. [CrossRef] [PubMed]
16. Kim, D.S.; Kim, S.W.; Song, J.M.; Kim, S.Y.; Kwon, K.C. A new prokaryotic expression vector for the expression of antimicrobial
peptide abaecin using SUMO fusion tag. BMC Biotechnol. 2019, 19, 13. [CrossRef] [PubMed]
17. Davis, G.D.; Elisee, C.; Newham, D.M.; Harrison, R.G. New fusion protein systems designed to give soluble expression in
Escherichia coli. Biotechnol. Bioeng. 1999, 65, 382–388. [CrossRef]
18. Li, J.; Xiao, W.; Yuan, D.; Wang, G.; Ma, L.X. Site-directed mutagenesis by combinantion of homologous recombination and DpnI
digestion of the plasmid template in Escherichia coli. Anal. Biochem. 2008, 373, 389–391. [CrossRef]
19. Wang, Y.P.; Rao, B.; Yan, H.; Han, R.; Li, L.; Liao, P.A.; Ma, L.X. High-level expression of l-glutamate oxidase in Pichia pastoris
using multi-copy expression strains and high cell density cultivation. Protein Expr. Purif. 2017, 129, 108–114.
20. Wang, Y.P.; Rao, B.; Zhang, L.; Ma, L.X. High-level expression of two thermophilic β-mannanases in Yarrowia lipolytica. Protein
Expr. Purif. 2017, 133, 1–7.You can also read

















































