Unique three-site compound heterozygous mutation in the WFS1 gene in Wolfram syndrome
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Ren et al. BMC Endocrine Disorders (2021) 21:166
https://doi.org/10.1186/s12902-021-00823-5
RESEARCH ARTICLE Open Access
Unique three-site compound heterozygous
mutation in the WFS1 gene in Wolfram
syndrome
Ziyu Ren1†, Jixiu Yi2†, Min Zhong3, Yunting Wang4, Qicong Liu1, Xuan Wang1, Dongfang Liu1* and Wei Ren4*
Abstract
Background: Wolfram syndrome (WFS) is a rare autosomal recessive genetic disease whose main cause is
mutations in the WFS1 and CISD2 genes. Its characteristic clinical manifestations are diabetes insipidus, diabetes
mellitus, optic atrophy and deafness.
Methods: In this study, two patients from this particular family underwent complete routine biochemical and
ophthalmic tests. Blood, urine, routine stool test, visual acuity (VA) examination, visual field assessment, funduscope,
optical coherence tomography and periorbital magnetic resonance imaging (MRI) scans were performed for each
patient to evaluate whether the nerve fiber layer around the optic nerve head was atrophied and next-generation
sequencing of target genes was performed in two patients.
Results: When the patients were diagnosed with Wolfram syndrome, their genetic analyses suggested unique
three-site compound heterozygous mutations (c.2314C > T + c.2194C > T + c.2171C > T) in exon 8 of both patients’
chromosome 4. One mutation (c.2314C > T) was a novel mutation in the known reports of Wolfram syndrome. As a
degenerative genetic disease, the types of gene mutations in the Chinese population are generally homozygous
mutations at the unit point or compound heterozygous mutations at two nucleotide change sites. However, the
two patients reported in this study are the first known cases of compound heterozygous mutations with three
mutation sites coexisting on the WFS1 gene in China or even globally.
Conclusions: This study expands the phenotypic spectrum of Wolfram syndrome and may reveal a novel mutation
pattern of pathogenesis of Wolfram syndrome. The implications of this discovery are valuable in the clinical
diagnosis, prognosis, and treatment of patients with WFS1.
Keywords: Wolfram syndrome, Diabetes mellitus, WFS1, Compound heterozygous mutation
* Correspondence: ldf023023@qq.com; sawakita0209@163.com
†
Ziyu Ren and Jixiu Yi contributed equally to this work.
1
Department of Endocrinology and Metabolism, The Second Affiliated
Hospital of Chongqing Medical University, No. 74, Linjiang Road, Yuzhong
District, Chongqing 400010, China
4
Department of Endocrinology and Metabolism, The First Affiliated Hospital
of Chongqing Medical University, No. 1, You-Yi Rd, Yu-zhong District,
Chongqing 400010, China
Full list of author information is available at the end of the article
© The Author(s). 2021 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License,
which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if
changes were made. The images or other third party material in this article are included in the article's Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons
licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain
permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the
data made available in this article, unless otherwise stated in a credit line to the data.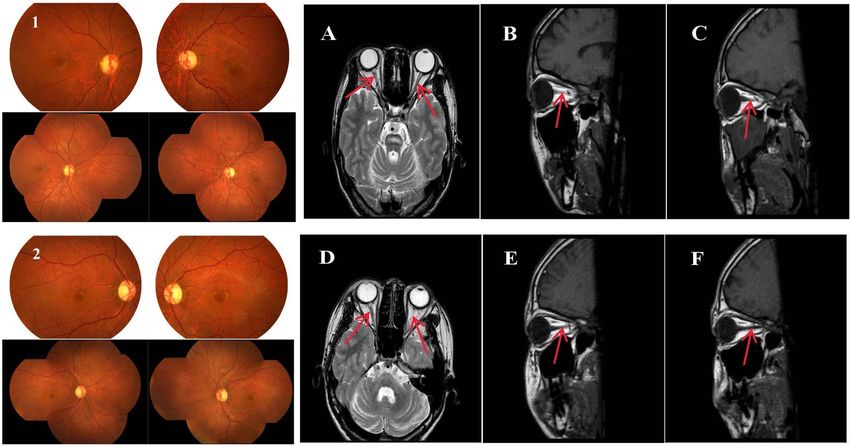
Ren et al. BMC Endocrine Disorders (2021) 21:166 Page 2 of 10
Background endoplasmic reticulum transmembrane protein that con-
WFS is an extremely rare autosomal recessive neurode- tains nine transmembrane segments and is embedded in
generative disorder first identified by Wolfram and the membrane in an Ncyt/Clum topology [12] and as-
Wagener in 1938 [1]. The prevalence of WFS is gener- sembles a high molecular weight complex of approxi-
ally extremely low, with a prevalence of 1 in 770,000 in mately 400 kDa in the membrane. It is widely
the UK [2], but is relatively high in certain parts of Asia, overexpressed in the brain, pancreas, heart, and insuli-
such as Lebanon, with a prevalence of 1 in 68,000 [3]. noma beta cell lines. Its main function may be the
The clinical manifestations of WFS are diverse and are homeostasis of Ca2 + regulated transport in protein syn-
mainly characterized by juvenile diabetes insipidus (DI), thesis modification [13]. The ERISP encoded by CISD2
diabetes mellitus (DM), optic atrophy (OA) and deafness is a new type of transmembrane protein located on the
(D). The term “DIDMOAD” is also used to describe pa- endoplasmic reticulum membrane. ERISP has been dem-
tients with WFS who have a variety of complications [4]. onstrated to have an effect similar to Wolframin in
Subsequent studies have shown that only 51% of patients maintaining endoplasmic reticulum and cell homeostasis
had signs of diabetes insipidus and deafness, and all four and Ca2+ transport [14].
features are seen in only 13% of patients [4]. However,
juvenile-onset diabetes mellitus and optic atrophy are al- Methods
ways present and appear first. Therefore, juvenile diabetes Study design
and optic atrophy are the best diagnostic criteria for Wol- In this study, we conducted a comprehensive clinical
fram syndrome [2, 5], and WFS is also classified as a spe- genetic investigation of a unique family case, systematic-
cial type of endocrine disease [6]. Because there is no ally reviewed the clinical ophthalmology and endocrin-
specific treatment, the prognosis of WFS is extremely ology characteristics of two patients (brothers) in this
poor. Patients mainly have multiple systems of progressive family, and performed genetic analysis of their immedi-
neurodegeneration; initial symptoms include color vision ate family members in the previous two generations. We
and peripheral vision loss; mild hearing loss and diabetes found that this case is different from the common
insipidus gradually develop into optic atrophy, deafness single-site homozygous mutation or double-site com-
and even ataxia, central nervous system dysfunction such pound heterozygous mutation type in that it is a rare
as central apnea, and eventually death [7]. three different site compound heterozygous mutation.
According to different pathogenic mechanisms, gener- We also identified 1 novel mutation in WFS1. Therefore,
alized WFS can be classified as Wolfram syndrome type in this article, we report the first case of a three-site
1 (WS1) and Wolfram syndrome type 2 (WS2) [2, 8]. compound heterozygous mutation in China and the
WS1 is characterized by a pathogenic gene on chromo- world.
some 4. WS2 was first reported in 2000 in four consan-
guineous Jordanian families [8]. In contrast to the typical Study participants
“DIDMOAD” symptoms of WF1, this new type of Wol- Patient 1
fram syndrome presents unique clinical manifestations: The proband, an 18-year-old boy (eldest brother), was
peptic ulcer disease, bleeding tendency secondary to a admitted to the hospital 9 years ago (9 years old) due to
platelet aggregation abnormality and absence of diabetes no obvious inducement of eye pain, haze, photophobia,
insipidus [9]. Follow-up studies have demonstrated that tears with thirst, polydipsia, polyuria, and nocturnal en-
the pathogenesis of WS2 is due to a CISD2 mutation on uresis. The initial diagnosis was “type 1 diabetes with
chromosome 4q22–24 [8]. The WFS1 gene and the double corneal ulcer”. After symptomatic treatment,
CISD2 gene participate in the separate regulation of two such as anti-inflammatory and insulin therapy to control
transmembrane proteins, “Wolframin” and “endoplasmic hyperglycemia, the patient was discharged from the hos-
reticulum interferon-stimulating protein” (ERISP), on pital and received long-term insulin injection treatment
the endoplasmic reticulum [10, 11]. Wolframin is an outside the hospital. Two years ago (16 years old), due to
Table 1 Clinical characteristics of patients with Wolfram syndrome (DIDMOAD)
Case Age Sex DI, age at DM, age at OA, age at HI, age at Other features, age at UCVA
no. diagnosis diagnosis diagnosis diagnosis diagnosis
1 18 M No Type 1, 9 years Bilateral, 16 years Left MF, 17 Years Abnormal MRI of brain, OD:
uroschesis 0.04
OS: 0.1
2 14 M No Type 1, 9 years Bilateral, 11 years No Abnormal MRI of brain, OD: 0.2
uroschesis OS: 0.1
DM diabetes mellitus, OA optic atrophy, DI diabetes insipidus, HI hearing impairment, MF medium-frequency hearing impairment, UCVA uncorrected visual acuity,
EEG Electroence phalography, OD right eye, OS left eye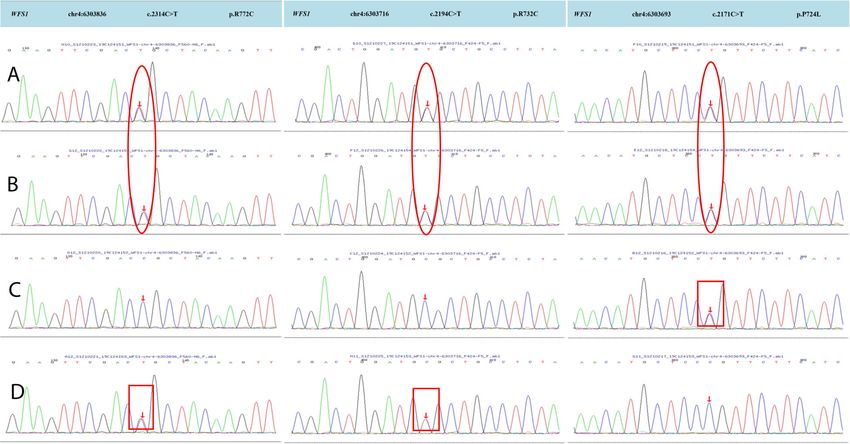
Ren et al. BMC Endocrine Disorders (2021) 21:166 Page 3 of 10
Table 2 Routine laboratory testing of patients with Wolfram syndrome
Case Endocrine TC Routine urine
no. routine test
HbA1c FBG C-P Cholesterol (mmol/ Triglyceride (mmol/ UGLU Urine ketone body Urine specific
% (mmol/ (mmol/L) (nmol/ L) L) gravity
mol) L)
1 12.9 15.01 0.07 5.22 1.09 ++++ Positive 1.029
2 8.4 10.79 0.05 4.71 0.87 +++ Positive 1021
HBA1c glycated hemoglobin, FBG fasting blood glucose, C-P c-peptide, TC triglyceride, UGLU urine glucose
the progressive decrease in binocular vision in the pa- administered for nearly 1 year. Two-plus years ago, there
tient, he was diagnosed in our hospital as “left eye optic was a progressive increase in blurred vision, frequent urin-
atrophy, left eye neurotrophic keratitis, and right eye ation, endless urination, and exacerbation of nocturnal en-
neurotrophic corneal ulcer”. The diagnosis and treat- uresis. He was admitted to local hospital with “diabetic
ment during this period are not quite clear. In recent ketoacidosis,” and physicians performed the relevant
years, the patient gradually felt hearing loss in the right examination (Table 1).
ear, accompanied by increased urinary incontinence at
night, so he was admitted to the hospital for further Laboratory analysis
treatment. Both patients underwent complete routine biochem-
ical and ophthalmic tests Including blood, urine, stool
Patient 2 routine test, VA examination, visual fields assessment
Another case in this family was a 14-year-old boy (youn- (Carl Zeiss Meditec, Inc., Dublin, CA, United States),
ger brother) who had symptoms similar to his elder broth- the funduscope, optical coherence tomography (Cirrus
er’s thirst, polydipsia, polyuria, and nocturnal enuresis OCT 5000, Carl Zeiss Meditec, Inc., Dublin, CA,
with blurred vision when he was 9 and was not treated. United States) and Periorbital MRI scans (philips
Three years ago (11 years old), his blood sugar increased, Achieva 1.5 T, Netherlands) was performed for each
so oral “Chinese medicine” anti-hyperglycemic was patient to evaluate the nerve fiber layer around the
Fig. 1 Fundus photography and periorbital magnetic resonance images of two patients. (1) Shows the fundoscopic results of patient 1, and (2) shows
the fundoscopic results of patient 2. Both figures show optic disc diffused pallid bilaterally without diabetic retinopathy. The two patients’ optic nerves
were pale, and the bilateral optic nerves were thinner. A-C Bilateral optic nerve atrophy from the coronal and sagittal positions in patient 1. D-F
Bilateral optic nerve atrophy from the coronal and sagittal positions in patient 2. Red arrows show the atrophic optic nerves of the two patients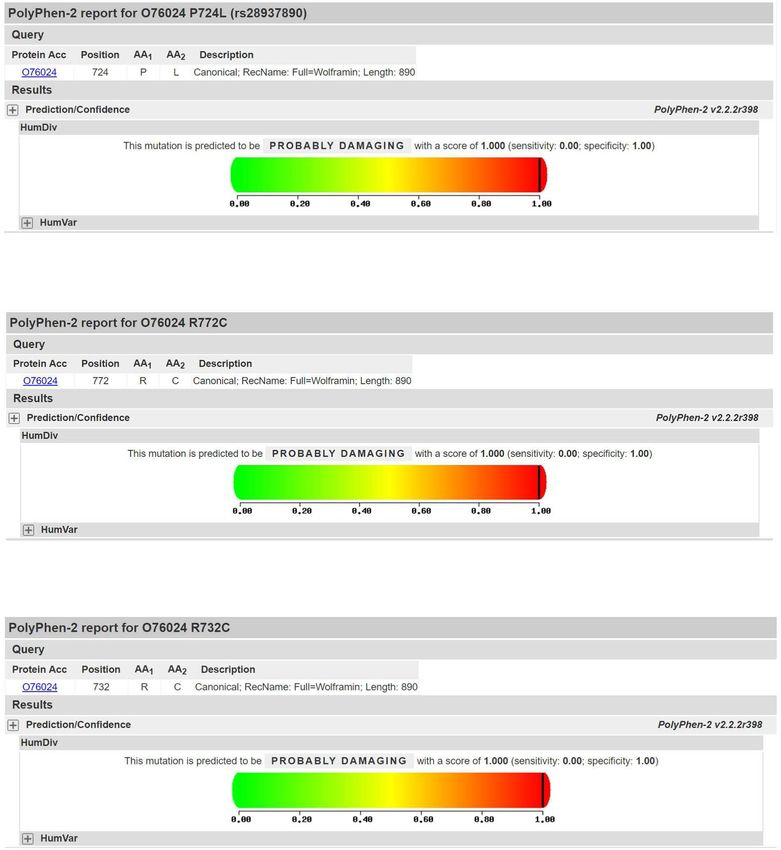
Ren et al. BMC Endocrine Disorders (2021) 21:166 Page 4 of 10
optic nerve head is atrophied. Brain MRI was per- Patient 2 (younger brother)
formed in 2 patients. The audiological, urological and Fasting blood glucose was 10.79 mmol/L, fasting c-
psychiatric examinations results were recorded from peptide was 0.33 nmol/L, HbA1c/Ghba1c was 8.4%, and
the medical records. he was urine sugar and urine ketone body positive.
Both patients had typical symptoms of thirst, poly-
dipsia, polyuria and weight loss, so they were diag-
Genetic analysis nosed with type 1 diabetes mellitus (DM1). The
Extract the whole blood and DNA samples of the two patient’s cholesterol, triglyceride, HDL cholesterol,
patients and their Family members with the permission LDL cholesterol and thyroid function were all normal.
of the patient guardian. Genetic testing of two patients Optic coherence tomography (OCT) was performed
with next-generation sequences (NGS), A panel includ- for each patient to evaluate retinal nerve fiber layer
ing 194 ophthalmology associated genes were sequenced (RNFL) thickness. Ophthalmic fundoscopy and perior-
by Illumina HiSeq 2000 (Illumina, Inc., San Diego, CA, bital MRI suggested that the ratio of the optic cup to
United States) sequencing system. The average depth the optic disc was unclear, the optic nerve was pale,
was 200x. Family members of the proband were vali- and the bilateral optic nerve was thinner, which are
dated by Sanger sequence. consistent with the diagnosis of OA (Figs. 1 and 2).
DM1 combined with OA meets the basic diagnostic
Results criteria of WFS. The pure tone listening test (PTA)
Some of the examination results for both patients are as revealed that patient 2 had moderate left conductive
follows and are presented in Table 2. All the clinical deafness. Brain MRI scans indicated that brainstem
details and images of the patients mentioned later were and cerebellar vermis atrophy were accompanied by
obtained with the consent of their parents. Patients and brainstem lamellar abnormal signal shadows in the
participants provided their written informed consent to two patients. No obvious abnormality was found by
participate in this study. chest radiograph and abdominal B-ultrasound.
Genetic analysis
Patient 1 (eldest brother, proband) Both patients had mutations in the WFS1 gene, and
Fasting blood glucose was 15.01 mmol/L, fasting c-peptide both had unique three-site compound heterozygous
was 0.07 nmol/L, and glycosylated hemoglobin (HbA1c/ mutations. No other gene mutations or mitochon-
Ghba1c) was 12.8%. drial genomic mutations were detected. Pedigrees of
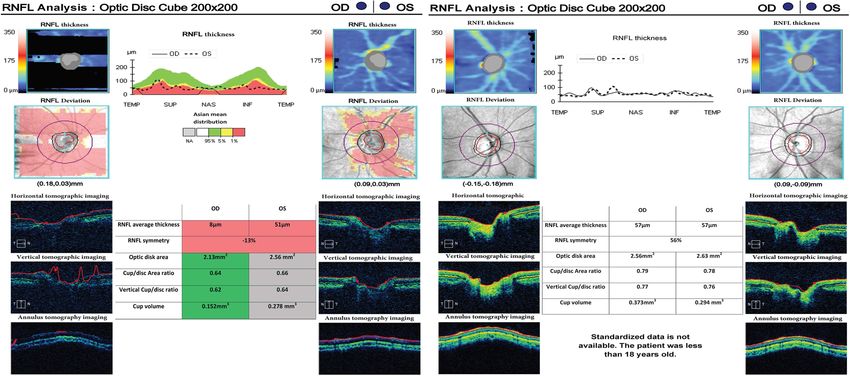
Fig. 2 Optic coherence tomography of two patients with WFS1 mutation. Optic coherence tomography (OCT) shows that peripapillary retinal
nerve fiber layer (RNFL) thickness significantly decreases. The left graph shows the thickness of the retinal nerve fiber layer in both eyes of patient
1. The right graph shows retinal thickness in both eyes of patient 2. The red part represents a decrease in thickness less than 1% outside normal,
and the green part indicates a decrease within the normal limit. Patient 2 represented in the right figure cannot be measured with standardized
data because he is under 18 years of age. RNFL: retinal nerve fiber layer; OD: right eye; OS: left eyeRen et al. BMC Endocrine Disorders (2021) 21:166 Page 5 of 10
this WFS family in our study are shown in Fig. 3. different nucleotide sites on the WFS1 and CSDI2
The mutation sites of these two patients were lo- genes. (Table 4).
cated in exon 8 of the WFS 1 gene c.2314C > T All papers are case reports on single nucleotide poly-
(p.R772C) + c.2194C > T (p.R732C) + c.2171C > T morphisms on the WFS1 and CISD2 genes in wolfram
(p.P724L) (Fig. 4). Both of these mutations were syndrome. According to available literature, patients
inherited from their maternal grandmother (Table 3). with wolfram syndrome are mostly female (21 female, 9
The c.2194C > T (p.R732C) and c.2314C > T male), all aged between 9 and 24 years old. The age of
(p.R772C) loci were searched using PP3 bioinformat- onset tends to be younger. Wolfram patients tend to
ics protein function prediction software, including show both severe juvenile diabetes and optic nerve at-
SIFT, PolyPhen_2, MutationTaster, GERP ++, rophy to varying degrees at a very early stage. The
REVEL, all of which indicate harmfulness (Fig. 5). disease is often diagnosed by genetic testing several
years after the first symptoms of DOMIO have pro-
gressively worsened. Among these 26 papers, five pa-
Literature review tients had wolfram syndrome caused by mutations in
We systematically identified all potentially relevant ar- the CISD2 gene, while the rest had disease caused by
ticles from PubMed and Web of Science, Search mutations at different nucleotide sites in exons 4, 5
terms about “Wolfram syndrome” and “WFS1” and and 8 of the WFS1 gene. Wolfram syndrome caused
“CISD2” and “gene mutation sites”, were used in vari- by the CISD2 gene mutation exhibits very different
ous combinations and permutations across the data- clinical features, characterized by peptic ulcer disease,
bases.There were a total of 26 papers. We selected bleeding tendencies secondary to abnormal platelet
case reports of only two types of wolfram syndrome, aggregation and absence of enuresis. Their survival
including cases with mutations at different nucleotide time was also longer compared to WFS1.
loci on two pathogenic genes between 2011 and 2019.
After screening and sorting, We eliminated wolfram Discussion
syndrome-related mechanistic studies and reviews, ex- In this study, we evaluated a unique family case. In
cluded duplicate loci reported in the literature, and fi- addition to the rare three-site compound mutation type,
nally selected 12 articles [11, 14–24]. All 12 articles c.2314C > T is the first reported new mutant site. Both
reported reports of disease caused by mutations at patients had diabetes as the first symptom at their early
Fig. 3 Pedigrees of this WFS family. Black squares: affected males; white squares: unaffected males; white circles: unaffected females; arrow: the probandRen et al. BMC Endocrine Disorders (2021) 21:166 Page 6 of 10
Fig. 4 High-throughput sequencing results of WFS1 in both patients and Sanger sequencing results of WFS1 in their parents. A, B, C, D The
genetic sequencing results of patient 1, patient 2, father, and mother, respectively. Patient 1 and patient 2 had all three heterozygous mutations
in exon 8 of the WFS1 gene. The father and mother of the patients had heterozygous mutations (c.2171C > T (p.P724L) and c.2314C > T
(p.R772C) + c.2194C > T (p.R732C), respectively. The red circle presents heterozygous mutations in the two patients. The red box presents
heterozygous mutations in their parents
stage accompanied by at least one of the characteristics apoptosis [5]. Changes in ER function lead to accu-
of WFS, which is consistent with the diagnostic criteria mulation of misfolded proteins and activation of the
of WFS [2]. During the progression of the disease, the unfolded protein response (UPR), a state known as
ophthalmic symptoms of these two patients appeared “ER stress” [26]. However, WFS1 plays a key role in
earlier and gradually worsened. Various ophthalmo- ER stress, and recessive or dominant mutations in
logical examinations show abnormalities (severe vision WFS1 consistently lead to neuronal and/or endocrine
loss, decreased color vision, narrow field of vision, ab- dysfunctions [27, 28]. According to existing reports,
normal visual evoked potential, etc.) that need to be dis- the mutation sites in the two genes are diverse, but
tinguished from diabetic retinopathy [25]. all of them are homozygous mutations and double-
Recently, studies have confirmed that the pathogen- site compound heterozygous mutations (Table 4).
esis of WFS is closely related to mutations in two However, the pathogenic pattern of this family is a
genes, WFS1 and CISD2. Where WFS1 regulates wol- unique “three-site” compound heterozygous mutation.
framin, a functional protein encoded in the endoplas- Mutations in the WFS1 gene in three generations of
mic reticulum (ER), which is involved in the patient’s family can be clearly seen in Fig. 4. The
posttranslational modification, folding, and assembly patients’ family pedigree shows that heterozygous mu-
of new synthetic proteins, such as insulin, calcium tations at two nucleotide change sites (c.2194C > T,
storage, REDOX regulation, steroid synthesis, and c.2314C > T) in the WSF1 gene have been present
Table 3 Sanger sequencing reveals WFS1 gene mutations and clinical manifestations in the patients’ maternal grandparents
Test Detection Detection Nucleotide Subject Clinical Results
gene location method changes manifestation
WFS1 chr4–6,303,836 Sanger c.2314C > T maternal grandfather negative No variation
sequencing
maternal negative Heterozygous
grandmother mutations
WFS1 chr4–6,303,716 Sanger c.2194C > T maternal grandfather negative No variation
sequencing
maternal negative Heterozygous
grandmother mutationsRen et al. BMC Endocrine Disorders (2021) 21:166 Page 7 of 10 Fig. 5 Protein function prediction for three mutant sites (c.2171C > T (p.P724L); c.2314C > T (p.R772C); c.2194C > T (p.R732C)) of Polyphen2 since the patients’ grandmother (older relatives were did not cause the disease. Through Sanger sequence unable to provide blood samples due to age issues). verification, the heterozygous mutation c.2171C > The prediction of protein function also suggested that T(p.P724L) in the WSF1 gene was identified in the the muations in the encoded at all three sites of the father, which was not causing disease. Unfortunately, protein were harmful. The conventional wisdom is two young male patients in this family underwent that this type of mutation should be pathogenic, but genetic selection and had three different nucleotide interestingly, the two-site compound heterozygous mutations inherited from their parents’ chromosomes mutation in the patient’s mother and grandmother at the same time. It is likely that this unprecedented
Ren et al. BMC Endocrine Disorders (2021) 21:166 Page 8 of 10
Table 4 Clinical mutated nucleotide site and patterns of patients in WFS1 and CISD2 genes of Wolfram syndrome
GENE Population Nucleotide changes Exon Zygosity References
WFS1 Polish c.1232 V > delGCTG Exon8 Homozygous
WFS1 Polish c. 1943G > A Exon8 compound heterozygote [15]
c. 2336 T > G
WFS1 Polish c. 1330C > G Exon8 Homozygous
WFS1 Iranian c.376G > A Exon8 homozygous [16]
WFS1 Iranian c.1672C > T Exon10 homozygous [17]
WFS1 Iranian c.330C > A Exon4 Homozygote [22]
WFS1 Turkish c.1832_11847del16 Exon 8 Compound heterozygote
c.1672C > T
Turkish c.1867delA Exon 8 Compound heterozygote [18]
c.1943G > A
Turkish c.376G > A Exon 4 Homozygote
WFS1 Chinese c.1760G > A Exon 8 Homozygote [19]
WFS1 Japanese p. N325_I328del heterozygote Homozygote [21]
CISD2 Chinese c.272_273del Exon 2 Homozygote [20]
CISD2 Moroccan c.215A > G Exon 2 Homozygote [14]
CISD2 Italian c.103 + 1G > A Intron 1 Homozygote [23]
CISD2 Caucasian Intragenic deletion Exon 2 Homozygote [24]
CISD2 Jordanian c.109G > C Exon 2 Homozygote [11]
WFS1 Chinese c.2314C > T Exon8 Compound heterozygote This study
c.2194C > T
c.2171C > T
“three-site” compound heterozygous mutation caused The mother of the two patients in this case is currently
the onset of WFS in the two young men. pregnant with her third child and has been pregnant for
It is worth noting that this type of mutation pattern in approximately 10 weeks. It is precisely because of the
WFS1 has never been reported in the literature. The unique pathogenic pattern and genetic mutation site of
nonpathogenic single-site heterozygous mutation and her family that we recommend close monitoring of the
the compound heterozygous mutations in the WFS1 child’s growth after birth, as well as appropriate screen-
gene on chromosome 4 of the parents were brought to- ing to ensure quality of life. This case also suggests that
gether on the same pair of chromosomes through gen- endocrinologists need to be vigilant in clinical work. The
etic recombination and eventually caused disease. This possibility of WFS should be considered in young DM
case likely reveals a new WFS pathogenic pattern. We patients with early vision loss. They should also be ad-
can boldly assume that the pathogenic ability of the het- vised to obtain a thorough prenatal test when their fam-
erozygous mutation site in the WFS1 gene may have a ily members are ready to become pregnant [29]. WFS as
“cumulative effect”. Mutational sites may acquire greater a special category of endocrine diseases [ICD-11] that
pathogenicity through multiple generations of genetic should receive more attention. We also strongly recom-
accumulation and eventually lead to disease. This hy- mend that patients with typical DM1 combined with
pothesis was confirmed in our study, in which two pa- ophthalmic symptoms undergo genetic testing to deter-
tients developed disease due to the simultaneous mine the disease type for subsequent treatment [30].
acquisition of both parental heterozygous mutation sites.
As WFS is an extremely complex neurodegenerative
genetic disease, its specific pathogenesis is not clear. At Conclusions
this stage of our study, there is a lack of subsequent Patients with juvenile onset diabetes with optic nerve at-
functional studies of genes and proteins. The mechanism rophy should be alerted to the possibility of WFS and
of the three-site mutation pattern also needs to be con- should undergo aggressive genetic analysis. Three-site
firmed by more case evidence and subsequent multidis- compound heterozygous mutation may be a potential
ciplinary and multiteam cooperation research. This is novel pathogenesis of WFS that deserves further in-
also the direction of this team’s follow-up research. depth study.Ren et al. BMC Endocrine Disorders (2021) 21:166 Page 9 of 10
Abbreviations Received: 16 September 2020 Accepted: 20 July 2021
WFS: Wolfram Syndrome; VA: Visual Acuity; MRI: Magnetic Resonance
Imaging; DI: Diabetes Insipidus; DM: Diabetes Mellitus; OA: Optic Atrophy;
ERISP: Endoplasmic Reticulum Interferon-stimulating Protein; NGS: Next-
generation Sequences; HbA1c/Ghba1c: Glycosylated Hemoglobin; DM1: Type References
1 Diabetes Mellitus; OCT: Optic coherence tomography; RNFL: Retinal nerve 1. Wolfram DJ. Diabetes mellitus and simple optic atrophy among siblings:
fiber layer; PTA: Pure tone listening test; UPR: Unfolded protein response; report of four cases. Mayo Clin Proc. 1938;9:715–8.
OD: Right eye; OS: Left eye; HI: Hearing impairment; MF: Medium-frequency 2. Barrett TG, Bundey SE, Macleod AF. Neurodegeneration and diabetes: UK
hearing impairment; UCVA: Uncorrected visual acuity; EEG: Electroence nationwide study of Wolfram (DIDMOAD) syndrome. Lancet. 1995;346(8988):
phalography; FBG: Fasting blood glucose; C-P: C-peptide; TC: Triglyceride; 1458–63. https://doi.org/10.1016/S0140-6736(95)92473-6.
UGLU: Urine glucose 3. Medlej R, Wasson J, Baz P, Azar S, Salti I, Loiselet J, et al. Diabetes mellitus
and optic atrophy: a study of Wolfram syndrome in the Lebanese
population. J Clin Endocrinol Metab. 2004;89(4):1656–61. https://doi.org/1
Acknowledgments
0.1210/jc.2002-030015.
We are thankful for the excellent technical assistance provided by the staff of
4. Gunn T, Bortolussi R, Little JM, Andermann F, Clarke Fraser F, Belmonte MM.
the Department of Endocrinology, The Second Affiliated Hospital and
Juvenile diabetes mellitus, optic atrophy, sensory nerve deafness, and
Affiliated Children’s Hospital of Chongqing Medical University. We are
diabetes insipidus-a syndrome. J Pediatr. 1976;89(4):565–70. https://doi.org/1
grateful for support from the Chongqing Key Laboratory of Translational
0.1016/S0022-3476(76)80387-3.
Medicine in Major Metabolic Diseases and The Qianjiang Central Hospital,
5. Ishihara H, Takeda S, Tamura A, Takahashi R, Yamaguchi S, Takei D, et al.
where we collect and process patients.
Disruption of the WFS1 gene in mice causes progressive beta-cell loss and
impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet.
Code availability 2004;13(11):1159–70. https://doi.org/10.1093/hmg/ddh125.
Not applicable. 6. International Classification of Diseases for Mortality and Morbidity Statistics,
11th Revision (Version: 04 / 2019). Available here https://icd.who.int/
Authors’ contributions browse11/l-m/en. Accessed 16 Apr 2020.
ZYR and JXY analyzed the data and wrote the manuscript. MZ, YTW, QCL 7. Zmyslowska A, Malkowski B, Fendler W, Borowiec M, Antosik K, Gnys P, et al.
and XW participated in data collection and analysis, and used bioinformatics Central nervous system PET-CT imaging reveals regional impairments in
software for relevant analysis and comparison. WR and DFL planned the pediatric patients with Wolfram syndrome. PLoS One. 2014;9(12):e115605.
study and guided experiments (ZYR and JXY have an equal contribution). https://doi.org/10.1371/journal.pone.0115605.
The authors read and approved the final manuscript. 8. El-Shanti H, Lidral AC, Jarrah N, et al. Homozygosity mapping identifies an
additional locus for Wolfram syndrome on chromosome 4q. Am J Hum
Funding Genet. 2000;66(4):1229–36. https://doi.org/10.1086/302858.
Not applicable. 9. Ajlouni K, Jarrah N, El-Khateeb M, et al. Wolfram syndrome: identification of
a phenotypic and genotypic variant from Jordan. Am J Med Genet. 2002;
Availability of data and materials 115:61–5.
The datasets generated and analysed during the current study are available 10. Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, Bernal-Mizrachi E, et al. A
in the [National Center for Biotechnology Information] repository, [https:// gene encoding a transmembrane protein is mutated in patients with
www.ncbi.nlm.nih.gov/gene/7466]. diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet. 1998;
The datasets generated and analysed during the current study are available 20(2):143–8. https://doi.org/10.1038/2441.
in the [Polymorphism Phenotyping v2] repository, [http://genetics.bwh. 11. Amr S, Heisey C, Zhang M, Xia XJ, Shows KH, Ajlouni K, et al. A homozygous
harvard.edu/pph2/]. mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram
syndrome 2. Am J Hum Genet. 2007;81(4):673–83. https://doi.org/10.1086/52
0961.
Declarations 12. Takeda K, Inoue H, Tanizawa Y, Matsuzaki Y, Oba J, Watanabe Y, et al. WFS1
(Wolfram syndrome 1) gene product: predominant subcellular localization to
Ethics approval and consent to participate endoplasmic reticulum in cultured cells and neuronal expression in rat brain.
All procedures performed in studies involving human participants were Hum Mol Genet. 2001;10(5):477–84. https://doi.org/10.1093/hmg/10.5.477.
reviewed and approved by the Ethics Committee of the second affiliated 13. Hofmann S, Philbrook C, Gerbitz KD, Bauer MF. Wolfram syndrome:
hospital of Chongqing medical university and with the 1964 Helsinki
structural and functional analyses of mutant and wild-type wolframin, the
declaration and its later amendments or comparable ethical standards. The WFS1 gene product. Hum Mol Genet. 2003;12(16):2003–12. https://doi.org/1
patients and participants provided their written informed consent to 0.1093/hmg/ddg214.
participate in this study. 14. Rouzier C, Moore D, Delorme C, Lacas-Gervais S, Ait-el-Mkadem S, Fragaki K,
et al. A novel CISD2 mutation associated with a classical Wolfram syndrome
Consent for publication phenotype alters Ca2+ homeostasis and ER-mitochondria interactions. Hum
We confirmed written informed consent was obtained from all individuals Mol Genet. 2017;26(9):1599–611. https://doi.org/10.1093/hmg/ddx060.
whose information is provided in this manuscript, as well as from the parent. 15. Zmyslowska A, Borowiec M, Antosik K, Szalecki M, Stefanski A, Iwaniszewska
Both parents of the patients had approved the use of the patient’s clinical B, et al. Wolfram syndrome in the polish population: novel mutations and
details and images. All the written informed consent was consent for the genotype-phenotype correlation. Clin Endocrinol. 2011;75:636–41. https://
publication of this manuscript. doi.org/10.1111/j.1365-2265.2011.04102.x.
16. Safarpour Lima B, Ghaedi H, Daftarian N, et al. c.376G>A mutation in WFS1
Competing interests gene causes Wolfram syndrome without deafness. Eur J Med Genet. 2016;
The authors declare no conflicts of interest. 59:65–9.
17. Bansal V, Boehm BO, Darvasi A. Identification of a missense variant in the
Author details WFS1 gene that causes a mild form of Wolfram syndrome and is associated
1
Department of Endocrinology and Metabolism, The Second Affiliated with risk for type 2 diabetes in Ashkenazi Jewish individuals. Diabetologia.
Hospital of Chongqing Medical University, No. 74, Linjiang Road, Yuzhong 2018;61(10):2180–8. https://doi.org/10.1007/s00125-018-4690-3.
District, Chongqing 400010, China. 2Department of Endocrinology and 18. Ustaoglu M, Onder F, Karapapak M, et al. Ophthalmic, systemic, and genetic
Metabolism, The Qianjiang Central Hospital, Chongqing, China. 3Department characteristics of patients with Wolfram syndrome. Eur J Ophthalmol. 2019;
of Neurological Disorders, Chongqing Medical University Affiliated Children’s 30:1099–105.
Hospital, Chongqing, China. 4Department of Endocrinology and Metabolism, 19. Li M, Liu J, Yi H, Xu L, Zhong X, Peng F. A novel mutation of WFS1 gene in
The First Affiliated Hospital of Chongqing Medical University, No. 1, You-Yi a Chinese patient with Wolfram syndrome: a case report. BMC Pediatr. 2018;
Rd, Yu-zhong District, Chongqing 400010, China. 18(1):116. https://doi.org/10.1186/s12887-018-1091-1.Ren et al. BMC Endocrine Disorders (2021) 21:166 Page 10 of 10
20. Zhang Y, Feng L, Kong X, Wu J, Chen Y, Tian G. Novel mutations and the
ophthalmologic characters in Chinese patients with Wolfram syndrome.
Orphanet J Rare Dis. 2019;14(1):190. https://doi.org/10.1186/s13023-019-11
61-y.
21. Morikawa S, Tajima T, Nakamura A, Ishizu K, Ariga T. A novel heterozygous
mutation of the WFS1 gene leading to constitutive endoplasmic reticulum
stress is the cause of Wolfram syndrome. Pediatr Diabetes. 2017;18(8):934–
41. https://doi.org/10.1111/pedi.12513.
22. Noorian S, Savad S, Mohammadi DS. A novel nonsense mutation in the
WFS1 gene causes the Wolfram syndrome. J Pediatr Endocrinol Metab.
2016;29(5):607–9. https://doi.org/10.1515/jpem-2015-0045.
23. Rondinelli M, Novara F, Calcaterra V, Zuffardi O, Genovese S. Wolfram
syndrome 2: a novel CISD2 mutation identified in Italian siblings. Acta
Diabetol. 2015;52(1):175–8. https://doi.org/10.1007/s00592-014-0648-1.
24. Mozzillo E, Delvecchio M, Carella M, et al. A novel CISD2 intragenic deletion,
optic neuropathy and platelet aggregation defect in Wolfram syndrome
type 2. BMC Med Genet. 2014;15:88.
25. Al-Till M, Jarrah NS, Ajlouni KM. Ophthalmologic findings in fifteen patients
with Wolfram syndrome. Eur J Ophthalmol. 2002;12(2):84–8. https://doi.
org/10.1177/112067210201200202.
26. Yamada T, Ishihara H, Tamura A, Takahashi R, Yamaguchi S, Takei D, et al.
WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle
progression and triggers the apoptotic pathway specifically in pancreatic
beta-cells. Hum Mol Genet. 2006;15(10):1600–9. https://doi.org/10.1093/
hmg/ddl081.
27. Cnop M, Toivonen S, Igoillo-Esteve M, Salpea P. Endoplasmic reticulum
stress and eIF2α phosphorylation: the Achilles heel of pancreatic β cells. Mol
Metab. 2017;6(9):1024–39. https://doi.org/10.1016/j.molmet.2017.06.001.
28. Mollereau B, Rzechorzek NM, Roussel BD, Sedru M, van den Brink DM, Bailly-
Maitre B, et al. Adaptive preconditioning in neurological diseases -
therapeutic insights from proteostatic perturbations. Brain Res. 2016;1648(Pt
B):603–16. https://doi.org/10.1016/j.brainres.2016.02.033.
29. de Heredia ML, Clèries R, Nunes V. Genotypic classification of patients with
Wolfram syndrome: insights into the natural history of the disease and
correlation with phenotype. Genet Med. 2013;15(7):497–506. https://doi.
org/10.1038/gim.2012.180.
30. Abreu D, Urano F. Current landscape of treatments for Wolfram syndrome.
Trends Pharmacol Sci. 2019;40(10):711–4. https://doi.org/10.1016/j.tips.2019.
07.011.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations.You can also read

















































