Time crystals: From Schr odinger to Sisyphus
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Time crystals: From Schrödinger to Sisyphus
arXiv:2109.06091v1 [cond-mat.other] 13 Sep 2021
Antti J. Niemi1, 2, 3, ∗
1
Nordita, Royal Institute of Technology, Stockholm University and Uppsala University,
Hannes Alfvéns väg 12, SE-106 91 Stockholm, Sweden
2
Pacific Quantum Center, Far Eastern Federal University, 690950 Sukhanova 8, Vladivostok, Russia
3
School of Physics, Beijing Institute of Technology,
Haidian District, Beijing 100081, People’s Republic of China
A Hamiltonian time crystal can emerge when a Noether symmetry is subject to a condition that
prevents the energy minimum from being a critical point of the Hamiltonian. A somewhat trivial
example is the Schrödinger equation of a harmonic oscillator. The Noether charge for its particle
number coincides with the square norm of the wave function, and the energy eigenvalue is a La-
grange multiplier for the condition that the wave function is properly normalized. A more elaborate
example is the Gross-Pitaevskii equation that models vortices in a cold atom Bose-Einstein conden-
sate. In an oblate, essentially two dimensional harmonic trap the energy minimum is a topologically
protected timecrystalline vortex that rotates around the trap center. Additional examples are con-
structed using coarse grained Hamiltonian models of closed molecular chains. When knotted, the
topology of a chain can support a time crystal. As a physical example, high precision all-atom
molecular dynamics is used to analyze an isolated cyclopropane molecule. The simulation reveals
that the molecular D3h symmetry becomes spontaneously broken. When the molecule is observed
with sufficiently long stroboscopic time steps it appears to rotate like a simple Hamiltonian time
crystal. When the length of the stroboscopic time step is decreased the rotational motion becomes
increasingly ratcheting and eventually it resembles the back-and-forth oscillations of Sisyphus dy-
namics. The stroboscopic rotation is entirely due to atomic level oscillatory shape changes, so that
cyclopropane is an example of a molecule that can rotate without angular momentum. Finally, the
article is concluded with a personal recollection how Frank’s and Betsy’s Stockholm journey started.
Based on talk at Frankfest, Högberga 2021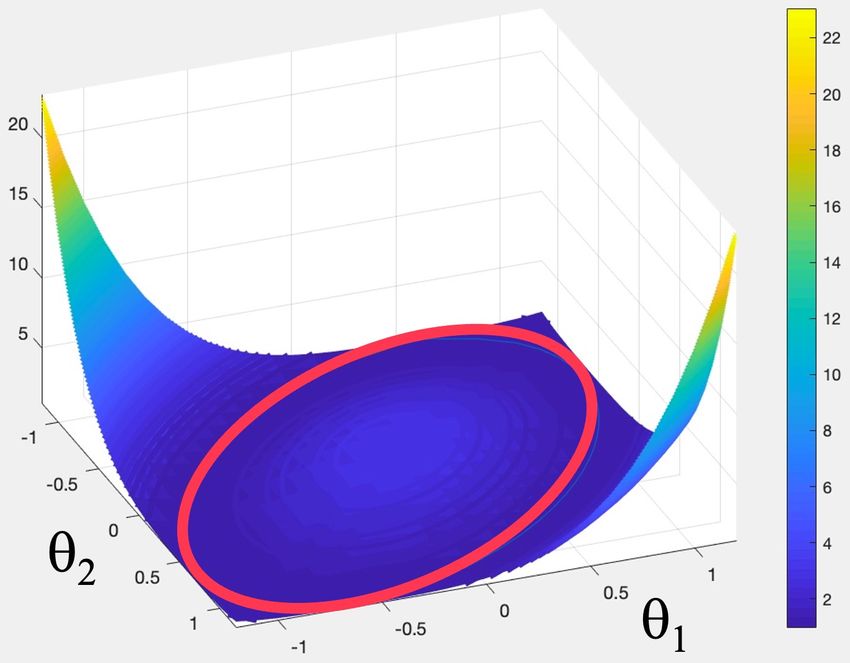
2
1. INTRODUCTION
Wilczek [1] together with Shapere [2] envisioned a time crystal to be a state that extends the notion of spon-
taneous breakdown of space translation symmetry to include time translation symmetry. They argued that if time
translation symmetry becomes spontaneously broken, the lowest energy state of a physical system can no longer be
time independent but has to change with time. If the symmetry breaking leaves behind a discrete group the ensuing
time evolution will be periodic, and in analogy with space crystals we have a time crystal.
The proposal drew immediate criticism: In a canonical Hamiltonian setting the generator of time translations is
the Hamiltonian itself. Thus energy is a conserved charge. But if time translation symmetry becomes spontaneously
broken, the ground state energy can no longer be a conserved quantity. Accordingly, a time crystal must be impossible
in any kind of isolated and energy conserving physical system, governed by autonomous Hamiltonian equation of
motion [3–5].
As a consequence the search of spontaneously broken time translation symmetry focused on driven, non-equilibrium
quantum systems [6–12]. The starting point is a many-body system with intrinsic dynamics governed by a time
independent Hamiltonian H. The system is then subjected to an external explicitly time periodic driving force, with
period T , that derives from an extrinsic time dependent potential U (t + T ) = U (t). The total Hamiltonian is the sum
of the two Htot (t) = H + U (t) so that the total Hamiltonian is also explicitly time dependent, with the same period
T of the drive. Floquet theory asserts that there can be solutions to the corresponding time dependent Schrödinger
equation, with a time period that is different from T where the difference is due to a Floquet index. As a consequence
it is plausible that in such a periodically driven many-body system, a spontaneous self-organization of time periodicity
takes place so that the system starts evolving with its own characteristic period, which is different from that of the
external driving force. It was first shown numerically that this kind of self-organisation can take place in certain
many-body localized spin systems [6–12]. Experiments were then performed in appropriate material realizations
[13–15] and they confirmed the presence of sustained collective oscillations with a time period that is indeed different
from the period of the external driving force. It is now widely accepted that this kind of driven non-equilibrium
Floquet time crystals do exist, but the setting is quite distant from Wilczek’s original idea.
Here I describe how, in spite of the No-Go arguments, genuine (semi)classical and quantum Hamiltonian time
crystals do exist [16–18]. They could even be widespread. I start with an explicit proof-of-concept construction
that shows how to go around the No-Go arguments: I show that whenever a Hamiltonian system supports conserved
Noether charges that are subject to appropriate conditions, the lowest energy ground state can be both time dependent
and have an energy value that is conserved. This can occur whenever the lowest energy ground state, as a consequence
of the conditions, is not a critical point of the Hamiltonian. I explain how such a time dependent ground state can be
explicitly constructed by the methods of constrained optimization [19, 20], using the Lagrange multiplier theorem [21].
Whenever the solution for any Lagrange multiplier is non-vanishing, the lowest energy ground state is time dependent:
A time crystal is simply a time dependent symmetry transformation that converts the time evolution of a Hamiltonian
system into an equivariant time evolution. In particular, since time translation symmetry is not spontaneously broken
but equivariantized, unlike in the case of conventional spontaneous symmetry breaking now there is no massless
Goldstone boson that can be associated with a time crystal. But the two concepts can become related in a limit
where the period of a time crystal goes to infinity and its energy approaches a (degenerate) critical point of the
Hamiltonian.
I present a number of examples, starting with the time dependent Schrödinger equation of a harmonic oscillator. I
continue with the Gross-Pitaevskii equation that models vortices in a cold atom Bose-Einstein condensate, and with a
generalized Landau-Lifschitz equation that can describe closed molecular chains in a coarse grained approximation. I
then analyze cyclopropane as an actual molecular example of timecrystalline Hamiltonian dynamics. For this I employ
high precision all-atom molecular dynamics to investigate the ground state properties of a single isolated cyclopropane
molecule. I conclude that the maximally symmetric configuration, with the D3h molecular symmetry, can become
spontaneously broken. I follow the time evolution of the minimum energy configuration stroboscopically, at very low
but constant internal temperature values. I find that with a proper internal temperature and for sufficiently long
stroboscopic time steps the molecule becomes a Hamiltonian time crystal with time evolution that is described by the
generalization of the Landau-Lifschitz equation. But when the length of the stroboscopic time step is decreased, there
is a cross-over transition to an increasingly ratcheting time evolution. In the limit where the stroboscopic time step
is very small the motion of the cyclopropane molecule resembles Sisyphus dynamics [22]. I propose that this kind
of cross-over transition between the Sisyphus dynamics that describes the limit of very short stroboscopic time steps,
and the uniform timecrystalline dynamics that describes the limit of very long stroboscopic time steps, is a universal
phenomenon. It exemplifies that the coarse graining of an apparently random microscopic many-body system can3
lead to a separation of scales and self-organization towards an effective theory Hamiltonian time crystal.
Finally, the rotational motion that I observe in a cyclopropane in the limit of long stroboscopic time steps, can
occur even with no angular momentum. Thus, cyclopropane is a molecular level example of a general phenomenon
of rotation by shape deformation, and without any angular momentum: The short time scale vibrational motions of
the individual atoms that are driven e.g. by thermal or maybe even by quantum mechanical zero point fluctuations
can become converted into a large scale rotational motion of the entire molecule, even when there is no angular
momentum. This kind of phenomenon was first predicted by Guichardet [23], and independently by Shapere and
Wilczek [24], and an early review can be found in [25].
2. HAMILTONIAN TIME CRYSTALS
A Hamiltonian time crystal describes the minimum of a Hamiltonian that is not a critical point of the Hamiltonian.
To show how this can occur, I start with the Hamiltonian action
Z
S = dt pi q̇i − H(p, q) (1)
where (pi , qi ) (i, j = 1, ..., N ) are the local Darboux coordinates with non-vanishing Poisson bracket
{qi , pj } = δij (2)
and Hamilton’s equation is
dqi ∂H
= {qi , H} =
dt ∂pi
(3)
dpi ∂H
= {pi , H} = −
dt ∂qi
On a compact closed manifold the minimum of H(p, q) is also its critical point, and in that case the Hamiltonian can
not support any time crystal [3–5]. Thus for a time crystal we need additional structure, and for this I focus on a
canonical Hamiltonian system that is subject to appropriate conditions
Ga (p, q) − ga = 0 a = 1, ..., n ≤ N (4)
where the ga are some constants and the Ga ’s define a Noether symmetry,
{Ga , Gb } = fabc Gc for all a, b, c
(5)
{Ga , H} = 0
I then search for a solution to the following problem [17]:
First, find a minimum of H(p, q) that is subject to appropriate conditions (4) but is not a
critical point of H(p, q). Then, solve (3) with this minimum as the initial condition.
The first step is a classic problem in constrained optimization [19, 20] and it can be solved using the Lagrange
multiplier theorem [21]. The theorem states that the minimum of H can be found as a critical point of
Hλ (p, q; λ) = H(p, q) + λa (Ga (p, q) − ga ) (6)
where the λa are independent, a priori time dependent auxiliary variables. The critical point of (6) is a solution of
∂H ∂Ga
+ λa = 0
∂pi ∂pi
∂H ∂Ga (7)
+ λa = 0
∂qi ∂qi
Ga (p, q) = ga
Since the number of equations (7) equals the number of unknowns (p, q, λ) a solution (p? , q ? , λ? ) including the Lagrange
multiplier, can be found at least in principle. Under proper conditions, in particular if H(p, q) is strictly convex and4
the Ga (p, q) are affine functions, existence and uniqueness theorems can also be derived. But if there are more than
one solution I choose the one with the smallest value of H(p, q).
Suppose the solutions λ?a for the Lagrange multipliers do not all vanish. By combining (7) with (3) I conclude that
I have a time crystal [17] with the initial condition
pi (t = 0) = p?i & qi (t = 0) = qi? & λa (t = 0) = λ?a (8)
and with the time evolution
dqi ∂Ga
= −λ?a = −λ?a {qi , Ga }
dt ∂pi
(9)
dpi ∂Ga
= λ?a = −λ?a {pi , Ga }
dt ∂qi
and the Lagrange multipliers can be shown to be time independent λa (t) ≡ λ?a . In particular, the timecrystalline
evolution (9) determines a time dependent symmetry transformation of the minimum energy configuration (8), one
that is generated by the linear combination
Gλa = λ?a Ga
of the Noether charges.
Since the Hamiltonian H has no explicit time dependence I immediately conclude that the energy of the time crystal
is a conserved quantity
dH
= −λ?a {H, Ga } = 0
dt
This contrasts some of the early arguments, to exclude a time crystal on the grounds that the minimum energy should
be time dependent.
In the sequel I consider exclusively such energy conserving Hamiltonian time crystals, with a time evolution that is
a symmetry transformation.
3. SCHRÖDINGER AS TIME CRYSTAL
For a simple example of the general formalism, I start with the following canonical action
Z
S = dtdx { ψ̄(i∂t )ψ − ψ̄(−∇2 + |x|2 )ψ } (10)
in D space dimensions. This action yields the non-vanishing Poisson bracket
{ψ̄(x1 ), ψ(x2 )} = −iδ(x1 − x2 ) (11)
The Hamiltonian energy is
Z
H = dx ψ̄(−∇2 + |x|2 )ψ (12)
and Hamilton’s equation coincides with the time dependent Schrödinger equation
i∂t ψ = −∇2 ψ + |x|2 ψ (13)
The Hamiltonian is strictly convex and its unique critical point is the absolute minimum
ψ(x) = 0
At this point I have an example of the situation governed by (3). In particular, there is no time crystal, as defined in
the previous Section.
To obtain a time crystal I follow the general formalism of the previous section: I introduce the square norm
Z
N = dx ψ̄ψ (14)5
This is the Noether charge for the symmetry of (10) under a phase rotation, it counts the number of particles, and I
subject it to the following familiar condition (4),
Z
G1 ≡ N − 1 = ( dx ψ̄ψ) − 1 = 0 (15)
I then proceed with the Lagrange multiplier theorem and search for the critical point of the corresponding functional
(6)
Z Z
2 2
HE = dx ψ̄(−∇ + |x| )ψ − E( dx ψ̄ψ − 1)
Where E ≡ −λ is the Lagrange multiplier: The corresponding equations (7) coincide with the time independent
Schrödinger equation for a harmonic oscillator
− ∇2 ψ + |x|2 ψ = Eψ
Z
dx ψ̄ψ = 1
In general there are many solutions, all the harmonic oscillator eigenstates are solutions. But I pick up the solution
? ?
(ψmin (x), Emin ) that minimizes the energy (12). This is exactly the textbook lowest energy ground state wave function
? ?
ψmin (x) of the D dimensional harmonic oscillator, and Emin is the corresponding lowest energy eigenvalue.
In line with (8), (9) I can write the time dependent Schrödinger equation (13) as follows,
Z
? ? ?
i∂t ψ = Emin {N, ψ} ≡ Emin { ψ̄ψ, ψ} with ψ(x, t = 0) = ψmin (x) (16)
That is, the time evolution of the harmonic oscillator wave function is a symmetry transformation generated by the
Noether charge N i.e. a phase rotation, with the familiar solution
?
ψ(x, t) = e−iEmin t ψmin
?
(x) (17)
Normally, a time dependent phase factor is not an observable. But it can be made so, e.g. in a proper double slit
experiment. Note that unlike in the case of standard spontaneous symmetry breaking, even though time translation
invariance is converted into an equivariant time translation, there is no Goldstone boson in a quantum harmonic
oscillator.
The previous example is verbatim a realization of the general formalism in Section 2. Albeit quite elementary
in its familiarity and simplicity, it nevertheless makes the point. Conditions such as (4) on Noether charges are
commonplace, they often have a pivotal role in specifying the physical scenario. When that happens, a time crystal
can appear. Moreover, without additional structure the appearance of a time crystal does not entail the emergence of
a massless Goldstone boson: A time crystal does not break the time translation symmetry. Instead, it equivariantizes
a time translation into a combination of a time translation and a symmetry transformation.
4. NONLINEAR SCHRÖDINGER AND TIME CRYSTALLINE VORTICES
I now proceed with additional examples of the general formalism. For this I observe that besides phase rotations
generated by the Noether charge N , the Schrödinger action (10) has also a Noether symmetry under space rotations.
Accordingly I introduce the corresponding Noether charges
Z
L = dx ψ̄(−ix ∧ ∇)ψ
Their Poisson brackets coincide with the Lie algebra SO(D) in D dimensions, and have vanishing Poisson brackets
with the conserved charge (14),
{L, N } = 0
I follow the general formalism: I impose additional conditions to the harmonic oscillator using the maximal commuting
subalgebra of space rotations; in three dimensions this amount to the familiar conditions L2 = l(l + 1) and Lz = m6
with l ∈ Z+ and m ∈ Z and |m| ≤ l. I can take results from any quantum mechanics textbook and confirm that all
the higher angular momentum eigenstates of the harmonic oscillator yield the appropriate minimum energy solutions
of the time dependent Schrödinger equation.
For a more elaborate structure, I introduce a quartic self-interaction and consider the following nonlinear Schrödinger
action,
Z
g
S = dtdx { ψ̄(i∂t )ψ − ψ̄(−∇2 + |x|2 )ψ − (ψ̄ψ)2 } (18)
2
This action also supports both N and L as conserved Noether charges. It defines the Gross-Pitaevskii model that
describes e.g. the Bose-Einstein condensation of cold alkali atoms at the level of a mean field theory [26], with g > 0
the quartic nonlinearity models short distance two-body repulsion. For clarity I specify to two space dimensions so
that there is only one conserved charge L ≡ Lz . The set-up is that of a spheroidal, highly oblate three dimensional
trap, with atoms confined to a disk by a very strong trap potential in the z-direction, and much weaker harmonic
trap potential in the (x, y) direction. Thus, besides the condition (15) I also introduce the following condition
Z
G2 ≡ Lz − lz = dxdy ψ̄(−ix × ∇)ψ − lz = 0 (19)
so that I have the scenario (4), (5) with two Noether charges.
I keep the normalization (15) but I leave the numerical value lz of the angular momentum as a free parameter.
The choice (15) is entirely for convenience: I can always rescale the square norm to set N to any non-vanishing value,
and compensate for this by adjusting the length and time scales. But lz and the parameter g in (18) remain as freely
adjustable parameters [27]. Note that even though the microscopic, individual atom angular momentum is certainly
quantized, the macroscopic angular momentum Lz does not need to be quantized: In applications to cold atoms the
wave function ψ(x, y) is a macroscopic condensate wave function that describes a very large number of atoms. Thus,
it can support arbitrary values of the macroscopic angular momentum Lz .
Following [27] I search for a solution of the nonlinear time dependent Schrödinger equation of (18) a.k.a. the
Gross-Pitaevskii equation
i∂t ψ = −∇2 ψ + |x|2 ψ + g|ψ|2 ψ (20)
that minimizes the Hamiltonian i.e. energy
Z
g
H = dx {ψ̄(−∇2 + |x|2 )ψ + (ψ̄ψ)2 } (21)
2
subject to the two conditions (15) and (19). The Hamiltonian is strictly convex, its global minimum coincides with
the only critical point ψ(x) ≡ 0. But when it is subject to the conditions (15) and (19) the minimum does not longer
need to be a critical point. Instead, the Lagrange multiplier theorem states that the minimum is a critical point
(ψ ? , λ?1 , λ?2 ) of
Hλ = H + λ1 G1 + λ2 G2 (22)
That is, the minimum is a solution of
δHλ
= −∇2 ψ + |x|2 ψ + g|ψ|2 ψ + λ1 ψ + λ2 (−ix × ∇)ψ = 0
δψ
Z
δHλ
= dx ψ̄ψ − 1 = 0 (23)
δλ1
Z
δHλ
= dx ψ̄(−ix × ∇)ψ − lz = 0
δλ2
If there are several solutions, I choose (ψ ? , λ?1 , λ?2 ) for which H(ψ) has the smallest value. The minimum energy ψ ? (x)
then defines the initial condition
ψ(x, t = 0) = ψ ? (x) (24)
for the putative timecrystalline solution of (20); recall that the corrresponding Lagrange multipliers are space-time
independent, λ1,2 (x, t) ≡ λ?1,2 .7
Remarkably, when I combine (23) and (20), the nonlinear time evolution equation of ψ ? (x) becomes converted to
the following linear time evolution equation
i∂t ψ = −λ?1 ψ + iλ?2 x × ∇ψ ≡ −λ?1 {G1 , ψ} − λ?2 {G2 , ψ} (25)
This is the equation (9) in the present case. In particular, (25) states that the time evolution of ψ ? (x) is a symmetry
transformation; the pertinent symmetry is a combination of phase rotation by N and a spatial rotation by Lz .
Since the Hamiltonian H has vanishing Poisson brackets with both G1 and G2 , the energy is conserved during the
timecrystalline evolution,
d
H = λ?1 {H, G1 } + λ?2 {H, G2 } = 0
dt
The solutions of (23), (20) describe vortices that rotate uniformly, with angular velocity −λ?2 around of the trap
center. For this I define
−1 x
Lz = −ix × ∇ & A = ∇ tan
y
to write (25) as follows:
λ?
i∂t ψ = −λ?2 Lz + 1? x × A ψ ≡ ωLcov
z ψ (26)
λ2
Here Lcov
z is the covariant angular momentum that generates the rotations around the trap center in the presence of
the ”analog gauge field” A. Note that A supports a ”magnetic” flux with ”strength” λ?1 /λ?2 along the z-axis, that
can have non-integer values. Notably, Lcovz has the same form as the angular momentum operator introduced by
Wilczek [28, 29], in the case of an anyon pair, in terms of the relative coordinate.
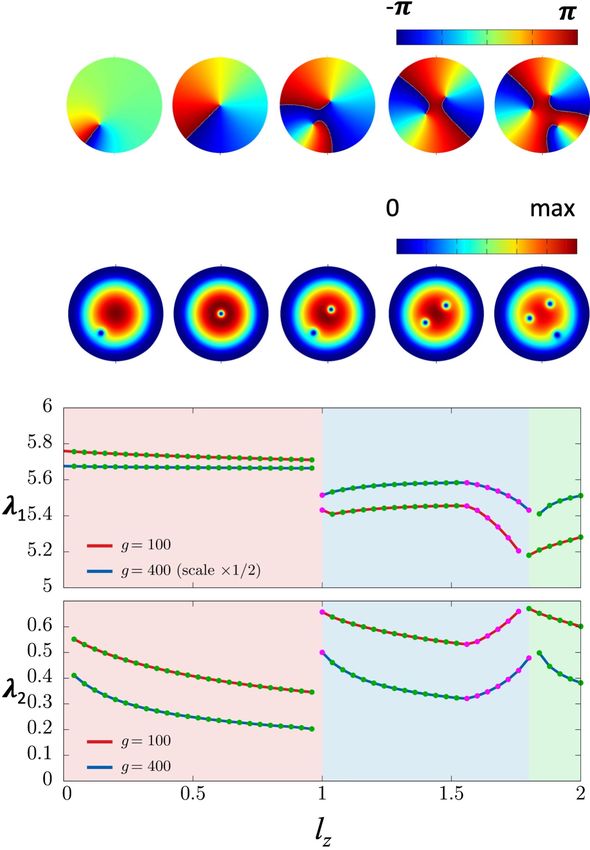
The Figure 1 summarizes the results from the numerical investigations in [ Garaud-2021a
¸ ]. The number of vortices
and their relative positions including the distances from the trap center, depend on the free parameter lz . The two
top lines of Figure 1 show how the phase and the cores of the vortex structure evolve for 0 < lz < 2. The bottom
panels show the corresponding values of the Lagrange multipliers λ?1 and λ?2 . Remarkably, when lz → 0 neither of
these multipliers vanish,
lim λ?1,2 (lz ) 6= 0 (27)
lz →0
Since λ?2 determines the angular velocity around the trap center (26), this means that for any lz 6= 0, 1 the minimum
energy solution describes a rotating vortex configuration.
For lz = 1 the vortex core coincides with the trap center, only its phase has time dependence. For lz = 0 the
minimum energy solution is a real valued function, up to a constant phase factor. For 0 < |lz | < 1 the distance
between the vortex core, located at point p ∈ R2 , and the trap center increases as |lz | decreases. Notably, the limit
lz → 0 is discontinuous and to inspect this I introduce the superfluid velocity
v(x, t) = ∇ arg[ψ](x, t) (28)
and I define its integer valued circulation
I
1
iv (p; Γ) = d` · v (29)
2π Γ
where Γ is a closed trajectory on the plane that does not go thru any singular point of v(x, t). For any given lz 6= 0
I can always choose Γ to be a circle around the trap center and with a large enough radius, so that the core p of a
given vortex is inside this circle. For a single vortex the value of (29) is iv (p; Γ) = ±1, with positive sign for lz > 0
and negative for lz < 0. For lz = 0 the value of the integral (29) vanishes, and for this value the phase of ψ(x, t) can
be chosen to vanish; the entire plane is a fixed point of (28) for lz = 0.
The circulation (29) is an integer valued topological invariant. In particular it can not be continuously deformed as
a function of lz , when lz varies between iv (p; Γ) = +1 for lz > 0 and iv (p; Γ) = −1 for lz < 0. When |lz | continues to
increase to values |lz | > 1 the value of (29) increases but always in integer steps, as new vortex cores enter inside the
(sufficiently large radius) circle Γ. Thus the vortex structures are topologically protected timecrystalline solutions of
(20).8 FIG. 1: The first line shows the phase of the wave function ψ ? (x, t). The second line shows the density |ψ ? (x, t)| for five representative instantaneous minimum energy vortex solutions of (20), (24): From left to right 0 < lz < 1.0, lz = 1, 0 < lz < 1.5, 1.5 < lz < 1.8, 1.8 < lz < 2.0; the numerical values 1.5, 1.8 are approximative, and depend on the coupling g. The vortices rotate around the trap center with angular velocity determined by λ?2 . In these figures the value of coupling is g = 400 which is representative for cold atom Bose-Einstein condensates [27]. The bottom panels show the evolution of Lagrange multipliers λ?1,2 for g = 100 and g = 400.
9
FIG. 2: The dynamical variables ni = ri+1 − ri are links that connect the vertices ri of a piecewise linear chain.
5. TIMECRYSTALLINE MOLECULAR CHAINS
I now proceed to a general class of Hamiltonian time crystals. The Hamiltonians describe discrete, piecewise linear
chains [16, 18]. One can think of the vertices ri (i = 1, ..., N ) as point-like interaction centers, they can e.g. model
atoms or small molecules in a coarse grained description of a linear polymer. The links ni = ri+1 − ri between the
vertices then model e.g. the covalent bonds, or peptide planes in the case of a protein chain. For clarity I only consider
cyclic chains, with the convention rN +1 = r1 .
In an actual molecule, the covalent bonds are very rigid and oscillate rapidly, with a characteristic period as short
as a few femtoseconds. I am mostly interested in timecrystalline dynamics at much longer time scales. Thus I assume
that the lengths of the links are constant, and equal to their time averaged values in the underlying atomistic system.
For simplicity I take all the links to have an equal length with the numerical value
|ni | ≡ |ri+1 − ri | = 1 (30)
The link variables ni are the dynamical coordinates in my set-up. I subject them to the Lie-Poisson bracket [16]
{nai , nbj } = ±abc δij nci (31)
where I can choose either sign on the r.h.s. and for clarity I choose +-sign; the two signs are related by parity
i.e. change in direction of ni . The bracket is designed to generate all possible local motions of the chain except for
stretching and shrinking of its links,
{ni , nk · nk } = 0
for all pairs (i, k), so that (30) is preserved for all times, independently of the Hamiltonian details.
Remarkably, the same bracket (31) appears in Kirchhoff’s theory of a rigid body that moves in an unbounded
incompressible and irrotational fluid that is at rest at infinity [30].
Hamilton’s equation coincides with the Landau-Lifschitz equation
∂ni ∂H
= {ni , H} = ni × (32)
∂t ∂ni
and the condition that the chain is closed gives rise to the first class constraints
N
X
G≡ ni = 0
i=1
(33)
{G , G } = abc Gc
a b
{G, H} = 0
The last equation restricts the form of Hamiltonian functions I consider. Such Hamiltonians Hi (n) can be constructed10
e.g. as linear combinations of the Hamiltonians that appear in the integrable hierarchy of the Heisenberg chain:
X
H1 = ai ni · ni+1
i
X
H2 = bi ni · (ni−1 × ni+1 )
i
X
H3 = ci ni · (ni−1 × (ni+1 × ni+2 )) (34)
i
X
H4 = di ni−1 · ni+1
i
etc.
where ai , bi , ci , di are parameters. Furthermore,
1
ri − rj = (nj + ... + ni−1 − ni − ... − nj−1 ) (35)
2
where I have symmetrized the distance, since there are two ways to connect ri and rj along the chain. As a consequence
I can also introduce two-body interactions between distant vertices as contributions in a Hamiltonian, such as a
combination of electromagnetic Coulomb potential and the Lennard-Jones potential:
N N
( 12 6 )
1 X ei ej X σP σvdW
U (x1 , ..., xN ) = + − (36)
2 i,j=1 |xi − xj | 2 i,j=1 |xi − xj | |xi − xj |
i6=j i6=j
Here ei is the charge at the vertex xi , σP characterizes the extent of the Pauli exclusion that prevents chain crossing,
and σvdW is the range of the van der Waals interaction. All are commonplace in molecular modeling, and in particular
they comply with the conditions (33).
Thus, the combination of (34) and (36) can be employed to construct realistic coarse grained Hamiltonian functions
H that describe dynamics of (closed) molecular chains, in a way that only depends on the vectors ni .
Note in particular that (33) implies that an initially closed chain remains a closed chain during the time evolution
(32).
I follow the general formalism of Section 2 to search for a time crystal as a minimum of H(n) that is subject to the
chain closure condition (33). The minimum is a critical point of the following version of (7), (9)
Hλ = H + λ · G
∂H
= −λ? & G(n? ) = 0
∂ni |n? (37)
∂H
⇒ = λ? × ni & ni (t = 0) = n?i
∂t
Whenever the solution λ? 6= 0 I have a time crystal as a closed polygonal chain that rotates like a rigid body. The
direction of its rotation and its angular velocity are both determined by the direction and the magnitude of the
time independent vector λ? . Thus, the present timecrystalline Hamiltonian framework provides an effective theory
framework for modeling the autonomous dynamics of rotating cyclic molecules.
In practice, to construct the minimum of H(n) I extend (32) to the Landau-Lifschitz-Gilbert equation for Hλ [17, 18]
∂ni ∂Hλ ∂Hλ
= −ni × + µ ni × (ni × ) (38)
∂t ∂ni ∂ni
with µ > 0 the large−t limit is a critical point of Hλ since (38) gives
N 2
dHλ µ X ∂ni
=− (39)
dt 1 + µ2 i=1 ∂t
so that the time evolution (38) proceeds towards decreasing values of Hλ and the flow continues until a critical point
(n?i , λ? ) is reached. Whenever more than one solution exist, I choose the one with smallest value of H.11
FIG. 3: a): A timecrystalline triangle with b = 0 and rotation axis determined by the ai . b): A timecrystalline
triangle with ai = 0, b 6= 0 and the rotation axis is the symmetric normal to the triangular plane. c): For generic
(ai , b) the rotation axis points to generic direction.
Simple examples
5.1 Triangular time crystal
The first simple example is an equilateral triangle, with Hamiltonian [16]
3
X
H= ai ni · ni+1 + bn1 · (n2 × n3 ) (40)
i=1
Hamilton’s equation (32) gives
∂t n1 = n1 × (a1 n2 + a3 n3 ) + bn1 × (n2 × n3 )
∂t n2 = n2 × (a2 n3 + a1 n1 ) + bn2 × (n3 × n1 ) (41)
∂t n3 = n3 × (a3 n1 + a2 n2 ) + bn3 × (n1 × n2 )
By summing these equations I obtain
d
(n1 + n2 + n3 ) = 0
dt
This confirms that an initially closed chain remains closed for all t. Moreover, for general choice of parameters (ai , b)
the r.h.s. of (41) never vanishes; the minimum of H for a closed chain is not a critical point of H; the minimum is
time dependent i.e. a time crystal. The time crystal describes rotation with an angular velocity around an axis, with
direction determined by the parameters. See Figure 3.
5.2 Knotted time crystals
The second concrete, simple example involves only the long range energy function (36), with Hamiltonian [18]
12
( 12 )
1X 1 3/4
H= + (42)
2 i,j=1 |ri − rj | |ri − rj |
i6=j
The first term is a Coulomb repulsion between the vertices and the second term is a short range Pauli repulsion that
prevents chain crossing; in an actual molecule the covalent bonds can not cross each other. The links connecting the
N = 12 vertices are chosen to have the topology of a trefoil knot. The initial knot geometry is otherwise random.
The Hamiltonian is first minimized using the Landau-Lifschitz-Gilbert equation (38). The set-up is an example of
(32), (33). The Figure 4 shows the resulting minimum energy time crystal, how it rotates according to (32) around
the axis that coincides with the vector λ? .
Various combinations of the local angles (34) and the long distance interactions (36) can be introduced. The ensuing
energy functions can describe time crystalline structures, also with more elaborate knot topologies than a trefoil. At a12
FIG. 4: A topological time crystal, with the topology of a trefoil knot, viewed from three different directions. The
energy function is given by (42), the rotation axis points in the direction of λ? and the angular velocity is
proportional to |λ? |.
higher level of realism, the interaction centers at the vertices can be given their internal atomic structure. Eventually,
one ends up with a highly realistic all-atom molecular dynamics description of a polymer chain, which is the subject
of the next Section.
6. CYCLOPROPANE AND SISYPHUS
6.1 Cyclopropane as a time crystal
All-atom molecular dynamics simulations are the most realistic descriptions that are presently available to model
chain-like molecules. CHARMM [31] is an example of such a molecular dynamics energy function, and GROMACS [32]
is a user-friendly package for performing simulations. A typical molecular dynamics energy function contains the
following terms; here the summations cover all atoms of the molecule and can also extend e.g. over ambient water
molecules.
X X X
V (xi ) = kbi (li − li0 )2 + kai (θi − θi0 )2 + Vin [1 − cos(nωi − γi )]
bonds angles torsions
( " 12 6 #) (43)
X ei ej ij rmin rmin
+ + −2
|xi − xj | 2 |xi − xj | |xi − xj |
i6=j
The first term describes the stretching and shrinking of covalent bonds; it is not present in (34), (36) where the Lie-
Poisson bracket (31) preserves the bond lengths. The second and third terms account for the bending and twisting in
the covalent bonds. These terms are akin quadratic/harmonic approximations to non-linear terms that are listed in
(34). The last term is a combination of the electromagnetic and Lennard-Jones interactions (36). There can also be
additional terms such as the Urey-Bradley interaction between atoms separated by two bonds (1,3 interaction), and
improper dihedral terms for chirality and planarity; these can also be descrobed by the higher order terms in (34).
The time evolution is always computed from Newton’s equation, with the force field that derives from (43).
In the case of a molecular chain, a typical characteristic time scale for a covalent bond oscillation that is due to
stretching and shrinking of the bond described by the first term in (43), can be as short as a few femtoseconds. In
practical observations of molecular motions the time scales are usually much longer, and the observed covalent bond
lengths commonly correspond to time averaged values. For the bending and twisting motions the characteristic time
scales are much longer than for stretching and shrinking. Thus a separation of scales should take place so that an
effective theory description becomes practical. Indeed, many phenomena that are duly consequences of the free energy
(43) can often be adequately modeled by an effective theory description. The effective theory energy function can be
a combination of terms such as those in (34), (36) and its dynamics can be described by the Lie-Poisson bracket (31),
in a useful approximation.
To investigate how the separation of scales takes place, and how an effective theory description emerges, in [ 33] all-
atom molecular dynamics has been used to simulate the ground state of an isolated cyclopropane molecule C3 H6 shown
in Figure ??. The force field is CHARMM36m, in combination with GROMACS. The simulation starts with a search of
the13
minimum energy configuration, at a given ultra low internal tem-
perature factor value; very low temperature thermal oscillations
could be interpreted as mimicking quantum oscillations in a semi-
classical description. The initial atomic coordinates can be found
from the PubMed site [34] where the positions of the carbon and
hydrogen atoms are specified with 10−14 m precision. In [ 33] the
structure is further optimized so that it describes a local minimum
of the CHARMM36m energy function, with full D3h molecular
symmetry where the carbon-carbon bond angles are 60o and the
hydrogen pairs are in full eclipse. Starting from this energy min-
imum all-atom trajectories are constructed, to simulate 10µs of
cyclopropane dynamics, at different ultra low internal tempera-
FIG. 5: Cyclopropane C3 H6 with D3h molecular ture factor values. The simulations use double precision floating
symmetry. point accuracy and the length of the simulation time step is 1.0f s;
for the detailed set-up and for simulation details I refer to [ 33].
All the individual atom coordinates are followed and recorded, and
analysed at different stroboscopic time steps ∆s t, during the entire 10 µs molecular dynamics trajectory.
When the cyclopropane is simulated with the very low ∼ 0.067K internal temperature factor value, and the all the
atom positions are observed at every ∆s t = 100ns (or longer) stroboscopic time step, the molecule rotates uniformly
at constant angular velocity. The axis of rotation coincides with the (time averaged) center of mass axis that is normal
to the plane of the three carbon atoms. Remarkably, this stroboscopic rotational motion is identical to the motion of
a triangular Hamiltonian time crystal shown in Figure 3 b): The dynamics is described by the generalized Landau-
Lifschitz equation (32), with the Hamiltonian H2 in (34. The results confirm that the timecrystalline Hamiltonian
H2 is an effective theory for this stroboscopic motion, in the limit of long stroboscopic time steps.
6.2 Spontaneous symmetry breaking
Notably, the Lie-Poisson bracket (31) breaks parity; if the sign on the r.h.s. is changed, the rotation direction
in Figure 3) b) also changes. Obviously something similar needs to take place in cyclopropane for it to rotate in a
particular direction: The cyclopropane is a priori a highly symmetric molecule with D3h molecular symmetry; the
carbon-carbon bond angles are all 60o and the dihedrals of all hydrogen pairs are fully eclipsed. In this maximally
symmetric state there can not be any unidirectional rotational motion around the molecular symmetry axis that
is normal to the plane of carbons, as there is no way to select between clockwise and counterclockwise rotational
direction. However, the bond angles are much smaller than the optimum 109.5o angles of a normal tetrahedral carbon
atom, and there is considerable torsional strain between the fully eclipsed hydrogen pairs [35]. Thus one can expect
that in the lowest energy ground state the D3h symmetry becomes spontaneously broken. By rigidity of covalent bond
lengths I expect that the symmetry breakdown should be mainly due to a twisting of the dihedral angles, between
the hydrogen pairs. This spontaneous symmetry breakdown selects a rotation direction, since parity is no longer a
symmetry. The simulation results that I have described show that this indeed occurs: The unidirectional rotation
around the triangular symmetry axis in the limit of long stroboscopic time steps is a manifestation of broken parity.
A simple model free energy can be introduced, to demonstrate how the spontaneous symmetry breaking due to
strain in hydrogen pair dihedral angles can take place. With θi (i = 1, 2, 3) the dihedrals, the free energy is
3
1X
F (θ1 , θ2 , θ3 ) = g(θi2 − α2 )4 (44)
4 i=1
The eclipsed configuration with all θi = 0 is a local maximum, and a critical point of the free energy. The minimum
of (44) occurs when θi = ±α, the value of α corresponds to the optimal value of the dihedral angle for two staggered
hydrogen pairs. But in the cyclopropane molecule the three dihedrals are subject to the condition
θ1 + θ2 + θ3 = 0 (45)
Thus θimin= ±α is not achievable for non-vanishing α. Instead I need to find the minimum of (44) subject to the
condition (45). This is (again) a problem in constrained optimization, so I search for critical points of
3
1X
Fλ = g(θi2 − α2 )4 + λ(θ1 + θ2 + θ3 )
4 i=114
with λ the Lagrange multiplier; I can rescale the angles, and set
a = g = 1 in which case the critical points obey
θi (θi2 − 1) + λ = 0 (46)
in addition of (45). I eliminate θ3 and plot the minimum of the
rescaled (44) as a function of θ1 and θ2 . The result is shown in
Figure 6. The minimum forms a circular curve on the (θ1 , θ2 )
plane around the origin. In particular, the D3h symmetry becomes
spontaneously broken, by the minimum. The Lagrange multiplier
can be solved from (46). It is non-vanishing except when θ1 has the
value 0 or ±1 and θ2 is ±1 or 0, respectively. The non-vanishing
of λ is suggestive of a time crystal, in line with the general theory
of Section 2.
FIG. 6: Free energy (44) with θ3 given by (45), and
its minimum.
6.3 Sisyphus
The Newtonian dynamics with the CHARMM36m force field is much more complex that the generalized Landau-
Lifschitz evolution (32) with the Hamiltonian H2 in (34). Thus one can not expect that the cyclopropane molecule
continues to display the same uniform, timecrystalline rotational motion when one decreases the length of the stro-
boscopic time step ∆s t. The Figure 7 shows how the rotational motion proceeds as function of the decreasing length
of the stroboscopic time step; these Figures display the instantaneous stroboscopic value of the rotation angle θ(t)
around the normal axis of the carbon triangle.
• Figure 7 a) shows the result for ∆s t = 100ns, when the effective theory description (32) with the Hamiltonian
H2 in (34) is accurate: The rotation is uniform, with a constant angular velocity.
• In Figure 7 b) the length of the stroboscopic time step is decreased to ∆s t = 20 ps. There is a very small amplitude
ratcheting, with an almost constant amplitude oscillations around the average value of the increasing rotation angle.
• In Figure 7 c) the stroboscopic time step is decreased to ∆s t = 2.0 ps. The amplitude of ratcheting oscillations
around the average value of θ has substantially increased: The molecule turns around regularly and rotates in the
opposite direction, but at a slightly smaller relative value of angular velocity ωs ≡ θ̇. The period of ratcheting
oscillations in θ(t) are also shorter than in Figure 7 b).
• Finally, in Figure 7 d) the molecule is sampled with stroboscopic time steps ∆s t = 200 f s. Now the motion is
becoming more chaotic, it consists of a superposition of very rapid back-and-forth rotations with different amplitudes
and frequencies, and with only a slow relative drift towards increasing values of θ(t).
The results shown in Figures 7 demonstrate how the separation of scales takes place in the simulated cyclopropane:
There is a continuous, smooth cross-over transition from a large-∆s t regime of a uniform timecrystalline rotation,
through an intermediate ∆s t regime with increasingly ratcheting rotational motion, to a small-∆s t regime that is
dominated by rapid back-and-forth oscillations, with different amplitudes and frequencies. Remarkably, when the
stroboscopic time step decreases, the time evolution of the cyclopropane becomes qualitatively increasingly similar
to the Sisyphus dynamics reported in [ 22]. Thus, the Sisyphus dynamics appears to provide a microscopic level
explanation how timecrystalline effective theory Hamiltonian dynamics emerges, at least in a small molecule such as
cyclopropane.
7. ROTATION WITHOUT ANGULAR MOMENTUM
Newton’s equation with the CHARMM36m all-atom molecular dynamics force field preserves the angular mo-
mentum, and angular momentum is also well preserved in a GROMACS numerical simulation. Since the initial
cyclopropane has no angular momentum, the rapid back-and-forth oscillations and in particular the uniform time-
crystalline rotational motion that is observed, are in apparent violation of angular momentum conservation. The
resolution of the paradox is that the cyclopropane is not a rigid body. It is a deformable body, and a deformable
body with at least three movable components can rotate simply by changing its shape [23, 24]. A falling cat is a good
example, how it can maneuver and rotate in air to land on its feet.15
FIG. 7: The evolution of the cyclopropane rotation angle during a 10 µs CHARMM36m simulation and with
T = 0.067K internal temperature factor value, recorded with decreasing stroboscopic time steps: a) The regime of
uniform rotation, here with ∆s t = 100 ns. b) and c) describe ratcheting regime, with ∆s t = 20 ps in b) and
∆s t = 2.0 ps in c). Finally d) with ∆s t = 200 f s is in the regime dominated by Sisyphus dynamics.
In the case of a cyclopropane molecule, when the internal temperature factor has a non-vanishing value, the covalent
bonds oscillate so that the shape of the molecule continuously changes; in an actual molecule there are also quantum
mechanical zero-point oscillations. Such shape changes are minuscule, but over a long trajectory their effects can
accumulate and self-organize into an apparent rotational motion. This is what is described in the Figures 7.
The analysis of results in [ 33] confirm that the angular momentum of the simulated cyclopropane is conserved,
and vanishes with numerical precision during the entire 10 µs simulation trajectory. For this, one evaluates the
accumulation of infinitesimal rotations, with each rotation corresponding to that of an instantaneous rigid body. An
infinitesimal rigid body rotation can be defined using e.g. Eckart frames, in terms of the instantaneous positions and
velocities of all the carbon and hydrogen atoms around the center of mass. In our simulations these are recorded
at every ∆τ = 10−15 f s time step n during the entire 10 µs production run. The instantaneous values L(n) of the
corresponding rigid body angular momentum component along the normal to the instantaneous plane of the three
carbon atoms can then be evaluated, together with the corresponding instantaneous moment of inertia values I(n).
This gives the following instantaneous “rigid body” angular velocity values
ω(n) = L(n)/I(n)
When these are summed up the result is the accumulated “rigid body” rotation angle, at each simulation step n:
n
X L(i)
ϑ(n) = ω(n)∆τ + ϑ(n − 1) = ∆τ (47)
i=1
I(i)
In full compliance with the conservation of angular momentum and the vanishing of its initial value, it is found [33]
that in all the 10 µs production run simulations the accumulated values (47) always remains less than ∼ 10−6 radians,
for all n, and the Figure 8 shows a typical example: There is no observable net rotation of the cyclopropane molecule
due to “rigid body” angular momentum, with numerical precision. Accordingly any systematic rotational motion that
exceeds ∼ 10−6 radians during a production run simulation must be emergent, and entirely due to shape deformations.
The original articles on rotation by shape deformations are [ 23, 24]. Reviews can be found in [ 18, 25] and here I
follow [ 18]: Consider the (time) t-evolution of three equal mass point particles that form the corners ri (i = 1, 2, 3)
of a triangle. I assume that there are no external forces so that the center of mass does not move,
r1 (t) + r2 (t) + r3 (t) = 0 (48)16
FIG. 8: The evolution of the rigid body rotation angle ϑ(n) in (47), during a typical cyclopropane simulation.
for all t. I also assume that the total angular momentum vanishes,
L = r1 × ṙ1 + r2 × ṙ2 + r3 × ṙ3 = 0 (49)
I now show that nevertheless, the triangle can rotate by changing its shape. To describe this rotational motion, I
place the triangle to the z = 0 plane and with the center of mass at the origin (x, y)=(0, 0). Two triangles then have
the same shape if they only differ by a rigid rotation on the plane, around the z-axis. I can describe shape changes
by shape coordinates si (t) that I assign to each vertex of the triangle. They describe all possible triangular shapes, in
an unambiguous fashion and in particular with no extrinsic rotational motion, when I demand that the vertex s1 (t)
always lies on the positive x-axis with s1x (t) > 0 and s1y (t) = 0, and the vertex s2 (t) has s2y (t) > 0 but s2x (t) can be
arbitrary. The coordinates s3 (t) of the third vertex are then fully determined by the demand that the center of mass
remains at the origin:
s3 (t) = −s1 (t) − s2 (t)
Now, let the shape of the triangle change arbitrarily, but in a T-periodic fashion. As a consequence the si (t) evolve
also in a T-periodic fashion,
si (t + T ) = si (t)
as the triangular shape traces a closed loop Γ in the space of all possible triangular shapes.
At each time t the shape coordinates si (t) and the space coordinates ri (t) are related by a rotation around the
z = 0 plane,
rix (t) cos θ(t) − sin θ(t) six (t)
= (50)
riy (t) sin θ(t) cos θ(t) siy (t)
Initially θ(0) = 0, but if there is any net rotation due to shape changes we have θ(T) 6= 0 so that the triangle has
rotated during the period, by an angle θ(T). I substitute (50) into (49) and I get
3
P
ZT ZT {siy ṡix − six ṡiy }
dθ(t) i=1
θ(T) ≡ dt = dt 3
(51)
dt
s2i
P
0 0
i=1
and in general this does not need to vanish, as I show in the next sub-section17
FIG. 9: The evolution of the angle θ(t) of (51) for the shape changes (52), and with stroboscopic time steps. In
panel a) ∆s t = 103 , In panel b) ∆s t = 102 , In panel c) ∆s t = 10, and in panel d) ∆s t = 1.
8. TOWARDS TIMECRYSTALLINE UNIVERSALITY
I now evaluate (51) in the case of a time dependent triangular structure, with three point-like interaction centers
at the corners. The shape changes are externally driven so that the shape coordinates evolve as follows:
1 cos[a sin ω2 t + 2π
1 cos[a sin ω1 t] 3 ]
s1 (t) = √ & s2 (t) = √ (52)
3 0 3 sin( 2π
3 )
I choose a = 0.1 and ω2 = 2ω1 = 3, substitute in (51) and evaluate the time integral numerically. The Figures 9 a)-d)
show how the angle θ(t) evolves, when observed with different stroboscopic time steps.
When I compare the Figures 7 and 9 I observe a striking qualitative similarity: Except for the scales, the corre-
sponding panels are almost identical. In particular, even though the shape changes (52) are externally driven, at large
stroboscopic time scales the time evolution again coincides with that of the autonomous Hamiltonian time crystal in
Figure 3 b).
The strong qualitative similarity between results in Figure 7 and 9 proposes that the Sisyphus dynamics of [ 22]
and the ensuing ratcheting at larger stroboscopic time scales, is akin a universal route, how an oscillatory short time
scale stroboscopic evolution becomes self-organized into a Hamiltonian time crystal, at large stroboscopic time scale.
Finally, when I expand the integrand (51), (52) in powers of the amplitude a, the result is
√
dθ 1 3 2 1
= − ω2 a cos ω2 t + a {ω2 sin 2ω2 t − ω1 sin 2ω1 t} − ω2 a3 cos 3ω2 t
dt 2 12 8
1
+ ω2 a3 {cos(ω2 + 2ω1 )t + cos(ω2 − 2ω1 )t} + O(a4 ) (53)
16
Thus, exactly when ω2 = ±2ω1 will the large time limit of the rotation angle θ(t) increase linearly in time as follows,
large−t 1
θ(t) −→ ω2 a3 t (54)
16
in line with Figure 9 a). But when ω2 6= ±2ω1 the large-T limit of the integral (51) vanishes, by Riemann’s lemma.
Similar high sensitivity of the rotation angle is also observed in the case of the cyclopropane, where the role of the
parameter a is played by the internal temperature factor of the molecule.18
CONCLUDING REMARKS
The concept of a time crystal has made a long journey. It started as a beautiful idea that was soon ridiculed as
a fantasy. From there it has progressed to the frontline of theoretical physics research, with high expectations for
remarkable future applications. But the notion is still very much under development. The conceptual principles are
still under debate and remain to be finalized, there are several parallel and alternative lines of research to follow. The
present article describes only my own personal way, how I try to understand what is a time crystal. My interest in
the subject started from discussions with Frank, and I am only able to describe what I have learned myself, by doing
things on my own, with Frank, and with my close colleagues. There is undoubtedly much that I have not covered,
but I leave it to others who know things better.
ACKNOWLEDGEMENTS
I thank Anton Alekseev, Jin Dai, Julien Garaud, Xubiao Peng and Frank Wilczek for collaboration on various
aspects of the original work described here. My research is supported by the Carl Trygger Foundation Grant CTS
18:276 and by the Swedish Research Council under Contract No. 2018-04411 and by Grant No. 0657-2020-0015 of
the Ministry of Science and Higher Education of Russia. I also acknowledge COST Action CA17139.
PERSONAL RECOLLECTIONS
I conclude with a short personal recollection of the remarkable way how Betsy and Frank have ended up, to spend
part of their time in Sweden and Stockholm University.
I first met Betsy and Frank personally in Aspen, during the summer 1984 session. Incidentally, we overlapped there
with Michael Green and John Schwarz, during the time when they gave birth to the modern string theory. At that
time Frank and I shared a more modest interest, and we discussed certain ideas around the SU(3) Skyrme model.
Unfortunately my self-confidence at the time was not at a level, to meet his challenges.
After Aspen we met several times, including Santa Barbara, Cambridge, Uppsala, Tours, Stockholm and elsewhere,
not to forget their lovely summer house in New Hampshire. Our discussions were always very enjoyable. Frank even
included me in his official delegation to attend the events during the Nobel Week, when he received the 2004 Physics
Prize.
In 2007, around the time when Nordita was moving to Stockholm, Frank told me that he had plans for a sabbatical.
I succeeded in attracting him to spend half a year in Sweden, jointly between Nordita and my Department at Uppsala
University. The other half of his sabbatical he spent at Oxford University, where I also visited him. There, he told me
about his dream to walk across the Great Britain. Apparently he hatched the first, very early ideas of a time crystal
during this walk. I now regret I did not ask to join him.
While in Uppsala, Frank and I got the idea to organize a Nobel Symposium in graphene. The Symposium took
place in Summer 2010, only a few months before the discovery of graphene was awarded the Physics Nobel Prize:
Ours was the first ever Nobel Symposium that took place the same year the Prize was awarded on the subject of a
Symposium. I understand that the many great talks, walks and discussions during the Symposium helped to decide
who got the Prize. Two years later, I hosted Frank when he became honorary doctor at Uppsala University. A little
later Frank came back to Uppsala, this time to collect Nobel chocolates that he won from a bet with Janet Conrad,
on the discovery of the Higgs boson.
Both Betsy and Frank seemed very happy with their sabbatical stay, and with all other experiences they had at
Stockholm and Uppsala. So I started to talk with Frank about the idea, that they could spend a little longer time in
Sweden, on a regular basis. In particular, I told Frank that the Swedish Research Council had occasional calls on a
funding program for International Recruitments, with very generous conditions. In 2014 the opportunity raised: Soon
after the Nobel Symposium on topological insulators at Högberga where Frank gave the summary talk, I learned that
the call was again being opened, and that this was probably the last call in the program. I contacted Frank, now with
more determination. He was at least lukewarm, so I proposed my colleagues at Uppsala University that we should
make an application to try and recruit Frank. Initially, I received several very positive, in fact some enthusiastic,
replies from my colleagues at the Department of Physics and Astronomy. But then came one strongly negative reply,
from Maxim Zabzine who at the time was responsible for the administration of theoretical physics. Maxim stated
that he did not want Uppsala University to make any effort, whatsoever, to apply for a Research Council grant for
Frank.19
I did not see any point to try and change Maxim’s strong opinion, in particular since the deadline for the appli-
cation was approaching. Instead, I immediately contacted Lars Bergström at Stockholm University. I asked him if
Stockholm University would be willing to submit an application to the Research Council, on Frank’s behalf. I reckon
that I contacted Lars only two days before the Nobel Prize of Physics 2014 was announced. Lars was at the time the
Permanent Secretary of the Nobel Physics Committee and for sure he had his hands full in preparing the announce-
ment. However, already the next day I received a reply from Anders Karlhede, then Vice President of Stockholm
University. Anders wrote that my proposal had been discussed with the President of Stockholm University Astrid
Söderberg Widding, and that Stockholm University is delighted to submit an application to the Research Council.
Soon after the Nobel Prize in Physics 2014 was announced and Lars became more free, he and Anders started to
work on the formal application; I understand they were also joined by Katherine Freese who was then Director of
Nordita. At that time I were in China with Frank. Frank was on his first-ever trip to China, and I coordinated the
visits in Beijing together with Vincent Liu. From Beijing we all continued to Hangzhou for the memorable inaugural
events of Wilczek Quantum Center at Zhejiang Institute of Technology.
During our travel in China I had several long discussions with Frank about the Research Council grant application.
He had his doubts, and I made my best to persuade him. I remember vividly the decisive discussion: After a breakfast
at our hotel, a historic place that used to be Mao Zedong’s favorite retreat at the West Lake in Hangzhou, Frank
and I were sitting together in the lobby. In Stockholm, the Research Council application was ready to be submitted,
the deadline was only a couple of days away. But Stockholm University still needed Frank to sign a formal letter of
interest for the application, and Frank was hesitant. However, after some lengthy discussion I got his signature, and
I sent the signed letter right away to Anders. In two months time we received a decision from the Research Council
that Stockholm University’s application for the International Recruitement Grant for Frank had been approved.
However, it was too early to call it a home run. For that, I still needed some advice from Anders Bárány, who
is the creator of Nobel Museum. Anders came up with the brilliant idea, of a curator position for Betsy at Nobel
Museum. To launch this, I teamed up with Gérard Mourou who invited Frank and Betsy, and a delegation from
Nobel Museum, to the Symposium on Fresnel, Art and Light at Louvre in Paris. Frank gave a beautiful talk, and
Betsy made contacts with Louvre art curators. With a strong support from Astrid and Anders things worked out
impressively for Betsy at the Nobel Museum, where her achievements included the extraordinary Feynman Exhibition
at ArtScience Museum of Marina Bay in Singapore 2018-2019 that she largely organized with Frank’s support and
great help from K K Phua. With some additional aid and support from here and there, including Grand hospitality
by Katherine Freese at a right time in summer 2015, and in particular with the consistent and very strong support
from Astrid and Anders, Betsy and Frank were finally convinced to try and start their present journey in Stockholm.
I am really grateful that I can follow and share so much of it with them.
∗
Antti.Niemi@su.se
[1] F. Wilczek, Phys. Rev. Lett. 109 160401 (2012)
[2] A. Shapere, F. Wilczek, Phys. Rev. Lett. 109 160402 (2012)
[3] P. Bruno, Phys. Rev. Lett. 111 070402 (2013)
[4] H. Watanabe, M. Oshikawa, Phys. Rev. Lett. 114 251603 (2015)
[5] N. Yamamoto, Phys. Rev. D92 085011 (2015)
[6] K. Sacha, Phys. Rev. A91 033617 (2015)
[7] K. Sacha, D. Delande, Phys. Rev. A94 023633 (2016)
[8] V. Khemani, A. Lazarides, R. Moessner, S.L. Sondhi, Phys. Rev. Lett. 116 250401 (2016)
[9] D.V. Else, C. Nayak, Phys. Rev. B93 201103 (2016)
[10] D.V. Else, B. Bauer, C. Nayak Phys. Rev. Lett. 117 090402 (2016)
[11] D. V. Else, B. Bauer, C. Nayak, Phys. Rev. X7 011026 (2017)
[12] N. Y. Yao, A. C. Potter, I.-D. Potirniche, A. Vishwanath, Phys. Rev. Lett. 118 030401 (2017)
[13] J. Zhang et.al. Nature 543 217 (2017)
[14] S. Choi et.al. Nature 543 221 (2017)
[15] J. Rovny, R. L. Blum, S. E. Barrett, Phys. Rev. Lett. 120 180603 (2018).
[16] J. Dai, A.J. Niemi, X. Peng, F. Wilczek Phys. Rev. A99 023425-9 (2019)
[17] A. Alekseev, J. Dai, A.J. Niemi JHEP 08 035 (2020)
[18] X. Peng, J. Dai, A.J. Niemi, New J. Phys. 22 085006 (2020)
[19] R. Fletcher, Practical Methods of Optimization. (Wiley, Chichester New York, 1987)
[20] J. Nocedal and S. J. Wright, Numerical Optimization, Springer Series in Operations Research. (Springer, Heidelberg, 1999)
[21] J.E. Marsden and T.S. Ratiu, Introduction to Mechanics and Symmetry A Basic Exposition of Classical Mechanical SystemsYou can also read

















































