The chrysanthemum lavandulifolium genome and the molecular mechanism underlying diverse capitulum types - Oxford Academic
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Horticulture Research, 2022, 9: uhab022
https://doi.org/10.1093/hr/uhab022
Article
The chrysanthemum lavandulifolium genome and the
molecular mechanism underlying diverse capitulum
types
Downloaded from https://academic.oup.com/hr/article/doi/10.1093/hr/uhab022/6510191 by guest on 04 March 2022
Xiaohui Wen1 , 2 ,† , Junzhuo Li1 ,† , Lili Wang3 ,† , Chenfei Lu1 , Qiang Gao2 , Peng Xu4 , 5 , Ya Pu1 , Qiuling Zhang1 , Yan Hong1 , Luo Hong1 , He Huang1 , Huaigen Xin3 ,
Xiaoyun Wu1 , Dongru Kang6 , Kang Gao1 , Yajun Li1 , Chaofeng Ma1 , Xuming Li3 , Hongkun Zheng3 , Zicheng Wang6 , *, Yuannian Jiao4 , 5 , *, Liangsheng Zhang2 , * and
Silan Dai1 , *
1 BeijingKey Laboratory of Ornamental Plants Germplasm Innovation & Molecular Breeding, National Engineering Research Center for Floriculture, Beijing
Laboratory of Urban and Rural Ecological Environment, Key Laboratory of Genetics and Breeding in Forest Trees and Ornamental Plants of the Ministry of
Education, School of Landscape Architecture, Beijing Forestry University, No. 35 East Qinghua Road, Beijing 100083, China
2 Genomics and Genetic Engineering Laboratory of Ornamental Plants, Department of Horticulture, College of Agriculture and Biotechnology, Zhejiang University,
No. 866 Yuhangtang Road, Hangzhou 310058, China
3 Biomarker Technologies Co., Ltd, No. 12 Fuqian Street, Shunyi District, Beijing 101300, China
4 State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, No. 20 Nanxincun, Beijing 100093, China
5 University of Chinese Academy of Sciences, No.19(A) Yuquan Road, Beijing 100049, China
6 State Key Laboratory of Crop Stress Adaptation and Improvement, Plant Germplasm Resources and Genetic Laboratory, Kaifeng Key Laboratory of
Chrysanthemum Biology, School of Life Sciences, School of Agriculture, Henan University, Jinming Road, Kaifeng 475004, China
*Corresponding authors. E-mail: wzc@henu.edu.cn; jiaoyn@ibcas.ac.cn; zls83@zju.edu.cn; silandai@sina.com
† These authors contributed equally
Abstract
Cultivated chrysanthemum (Chrysanthemum × morifolium Ramat.) is a beloved ornamental crop due to the diverse capitula types
among varieties, but the molecular mechanism of capitulum development remains unclear. Here, we report a 2.60 Gb chromosome-
scale reference genome of C. lavandulifolium, a wild Chrysanthemum species found in China, Korea and Japan. The evolutionary analysis
of the genome revealed that only recent tandem duplications occurred in the C. lavandulifolium genome after the shared whole genome
triplication (WGT) in Asteraceae. Based on the transcriptomic profiling of six important developmental stages of the radiate capitulum
in C. lavandulifolium, we found genes in the MADS-box, TCP, NAC and LOB gene families that were involved in disc and ray f loret
primordia differentiation. Notably, NAM and LOB30 homologs were specifically expressed in the radiate capitulum, suggesting their
pivotal roles in the genetic network of disc and ray f loret primordia differentiation in chrysanthemum. The present study not only
provides a high-quality reference genome of chrysanthemum but also provides insight into the molecular mechanism underlying the
diverse capitulum types in chrysanthemum.
Introduction types, which restricts the utilization of rich flower type
Cultivated chrysanthemum (Chrysanthemum × morifolium resources in chrysanthemum. To date, the genomes of 16
Ramat.) is a well-known ornamental crop showing very Asteraceae species have been sequenced, which provides
diverse f lower morphologies. The f lower of a chrysanthe- insights into the Asteraceae genome [2]. However, most
mum is actually a capitulum that comprises inner disc of these species have a relatively distant relationship
florets and peripheral ray f lorets. The f lower types of with chrysanthemum. The genome sequencing quality
chrysanthemum are determined by the morphology and of some Asteraceae species was relatively low due to
relative numbers of disc and ray f lorets on a capitulum the limitation of sequencing technology and the high
[1]. Understanding the molecular mechanism of disc heterozygosity of Asteraceae (Supplementary Table 1).
and ray f loret differentiation under the same genetic The genomes of C. seticuspe and C. nankingense, belonging
background will provide not only a foundation for to the genus Chrysanthemum, have been sequenced by
the clarification of complex capitulum morphology in the Illumina sequencing platform and Oxford Nanopore
chrysanthemum but also insight into the f loral devel- long-read technology, respectively, without reaching
opment mechanism in Asteraceae. However, studies on the chromosome level [3, 4]. Therefore, it is necessary
the mechanism of f lower type in chrysanthemum are to obtain a high-quality chromosome-scale genome
hindered by the complex background of chrysanthe- of the genus Chrysanthemum and study the origin of
mum, making it difficult to directionally breed for flower chrysanthemum at the whole-genome level.
Received: April 10, 2021; Accepted: September 17, 2021; Published: 20 January 2022
© The Author(s) 2022. Published by Oxford University Press on behalf of Nanjing Agricultural University. This is an Open Access article distributed under the terms
of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted reuse, distribution, and reproduction
in any medium, provided the original work is properly cited.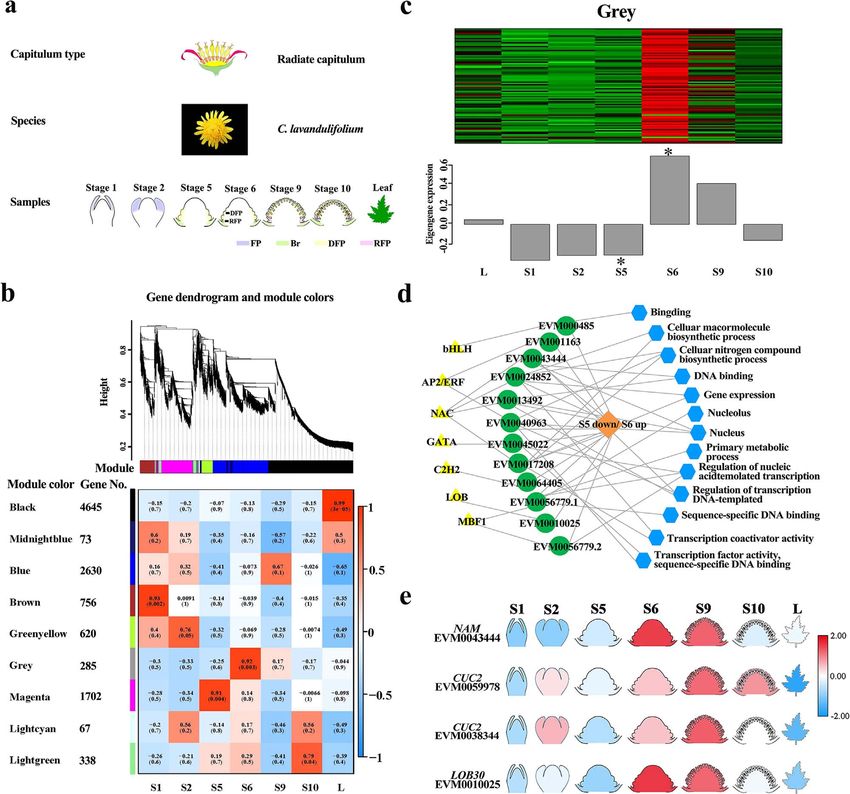
2 | Horticulture Research, 2022, 9: uhab022
Flower development is a complex process that involves Table 1. Assembly summary for the C. lavandulifolium genome at
the chromosomal level
a complex gene regulatory network [5, 6]. The expression
Assembly statistics Size/Number
patterns of abundant genes could be detected during
flower development using transcriptomic and genomic Contig number 10 136
sequencing technology. Studies on single f lowers have Contig length (bp) 2 669 472 274
Contig N50 (bp) 496 998
revealed that ABCE-class and CYC2-LIKE genes are
Contig N90 (bp) 136 070
involved in f loral organ identity and the regulation of Average length (bp) 15 002 629
floral symmetry [6, 7]. The development of next- and Maximum length (bp) 4 500 000
third-generation sequencing technologies enables the Minimum length (bp) 14 015
generation of high-quality genomes, and subsequently, GC% 36.02
Repeat 66.15%
these reference genome data can provide more infor-
Number of complete genes 64 257
mation to study the molecular mechanism of flower
Downloaded from https://academic.oup.com/hr/article/doi/10.1093/hr/uhab022/6510191 by guest on 04 March 2022
Scaffold number 178
development [8]. Previous studies showed that the Scaffold N50 (bp) 300 401 778
expression patterns and copy numbers of these genes Scaffold N90 (bp) 212 345 931
could regulate f loral organ morphology to modify flower Largest scaffold (bp) 322 278 347
Total scaffold size (bp) 2 670 468 074
types [5, 7, 9]. Comparative genomic analysis can also
help to reveal the copy numbers and evolution of floral
development-related genes [10–12]. To date, several tran-
scriptome sequencing profiles have been carried out in C. erozygosity) for C. lavandulifolium based on the results
lavandulifolium, C. nankingense and C. × morifolium “Jinba” from the K-mer distribution (Supplementary Figure 1).
[3, 13]. However, the analysis of these transcriptomes In total, 269.39 Gb (102.04 × coverage) and 193.88 Gb
lacked either a reference genome or the transcriptomic (62.50 × coverage) of data were generated using the
profiles of disc and ray f loret differentiation stages. PacBio RS II platform (Pacific Biosciences, Menlo Park,
Moreover, studies in Gerbera hybrida, Cosmos bipinnata CA, USA) and Oxford Nanopore sequencing technologies
and Senecio vulgaris found additional f loral-related (Oxford Nanopore Technologies Limited, Oxford Science
genes involved in capitulum development [14–16]. In Park, Oxford, UK), respectively. These sequences were
conclusion, more transcriptomes and reference genomes independently assembled by WTDBG v2.5 (https://gi
of chrysanthemum are needed to explore hub genes that thub.com/ruanjue/wtdbg), and the two draft genomes
participate in the molecular mechanism underlying the were merged and redundant reads were removed by
diverse capitula types. Quickmerge v0.3.021 and purge haplotigs v1.0.422 , which
The diploid species C. lavandulifolium (2n = 2x = 18) is resulted in a 3.10 Gb genome (Supplementary Table 2).
often regarded as one of the ancestral species of chrysan- With the aid of high-resolution chromosome confor-
themum [17, 18]. It is also used as a model plant to mation capture (Hi-C) technology, 2.93 Gb (94.46%) of
study the diverse capitula types in chrysanthemum due contigs were anchored onto the 9 chromosomes, forming
to its simple capitulum type, which possesses only one pseudomolecules (Figure 1 and Supplementary Table 3).
round peripheral ray f loret and many round inner disc After the removal of redundant and heterozygous
florets [19, 20]. Here, we sequenced and anchored 2.60 Gb sequences by the heatmap signal of Hi-C, a 2.60 Gb
sequences to 9 pseudochromosomes of C. lavandulifolium. chromosomal-level genome for C. lavandulifolium was
Phylogenetic analysis showed that C. lavandulifolium was obtained, with a contig N50 of up to 497 kb (Figure 1a,
an important donor species for chrysanthemum [17]. Table 1 and Supplementary Figure 2). Compared with the
Transcriptomic profiles of different developmental estimated C. lavandulifolium.
stages in radiate (C. lavandulifolium, Chrysanthemum genome size by K-mer distribution analysis, 98.48%
indicum, Chrysanthemum vesticum, C × morifolium “28”, of the C. lavandulifolium sequences were successfully
Erigeron brevisca, Helianthus annuus), discoid (Hippolytia assembled and anchored onto the nine chromosomes
alashanensis and Helenium aromaticum) and ligulate (Supplementary Table 3).
(Lactuca sativa and Taraxacum kok-saghyz) capitula This C. lavandulifolium genome is the first chromosome-
were also analyzed to explore the hub genes involved level genome in the genus Chrysanthemum. The protein-
in diverse capitulum development. Our study not only coding genes were annotated by ab initio prediction,
provides a high-quality reference for the assembly of homology and transcriptome-based approaches, and the
chrysanthemum genomes but also sheds light on the results were then integrated by Evidence Modeler. A total
regulatory mechanism of capitulum development in of 64 257 protein-coding genes were predicted (Supple-
Asteraceae. mentary Table 4), and the number of genes supported
by homology prediction and transcriptome prediction
was 53 714, accounting for 83.59% (Supplementary
Results Figure 3). Furthermore, 54 203 (84.35%) genes in the
Genome sequencing, assembly and annotation present reference genome were annotated by functional
The estimated genome size was approximately 2.64 databases (Supplementary Table 5). We identified 1417
Gb (with 68.57% repetitive sequences and 1.45% het- noncoding RNAs classified into 44 families: 16 families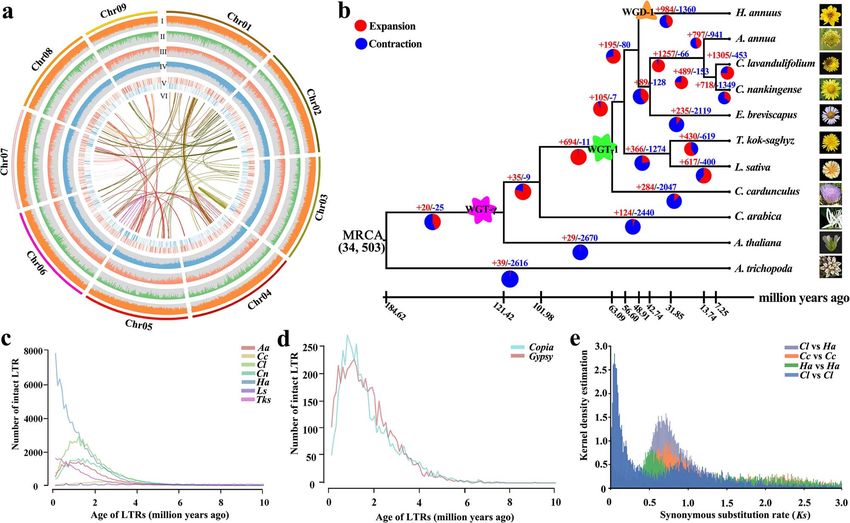
Wen et al | 3
Downloaded from https://academic.oup.com/hr/article/doi/10.1093/hr/uhab022/6510191 by guest on 04 March 2022
Figure 1. Overview and genome evolution of the C. lavandulifolium genome. a, Genomic features of the C. lavandulifolium genome and the expression
profile data during capitulum development. I, density of repetitive sequences; II, GC content; III, gene density; IV, heterozygosity; V, expression of
differentially expressed genes in stage 5 versus stage 6 of capitulum development (stage 5, the formation stage of disc f loret primordia, in which the
f loret primordia began to initiate; stage 6, the formation stage of ray f loret primordia, in which the disc and ray f loret primordia began to differentiate
on the capitulum); and VI, collinearity block between different pseudochromosomes. b, Phylogenetic gene tree of C. lavandulifolium with 10 other
plant species (Amborella trichopoda, Arabidopsis thaliana, Coffee arabica, Cynara cardunculus, Lactuca sativa, Taraxacum kok-saghyz, Helianthus
annuus, Artemisia annua, Erigeron breviscapus, C. nankingense, and C. lavandulifolium). c, Analysis of intact LTR numbers and insertion time in 7
Asteraceae plants. d, Analysis of Copia and Gypsy copy numbers in C. lavandulifolium. e, Synonymous substitution rate (Ks) distribution for pairs of
syntenic paralogs in C. lavandulifolium and two other plants (H. annuus and C. cardunculus).
of miRNA, 4 families of rRNA, and 24 families of tRNA Evolution of the C. lavandulifolium genome
(Supplementary Table 6). A total of 12 097 pseudogenes To study the conservation and specificity of the genomic
were identified using GeneWise. Benchmarking Universal structure of C. lavandulifolium, clustering of the predicted
Single-Copy Orthologs (BUSCOs) evaluation showed that proteins in the C. lavandulifolium genome with those from
89.02% and 92.36% of complete genes were obtained 4 other representative Asteraceae species showed 11 419
in genome mode and protein mode, respectively, which gene families shared by 5 species, with 2750 gene families
suggested the high quality of our assembled C. lavanduli- specific to C. lavandulifolium (Supplementary Figure 4).
folium genome (Supplementary Table 7). Gene Ontology (GO) enrichment analysis revealed that
Based on the high-quality reference genome in this C. lavandulifolium-specific genes were mainly enriched
study, 1.76 Gb of repetitive sequences of C. lavandulifolium in cellular process (GO: 0044763), metabolic process
were predicted, with 60.25% being retrotransposons and (GO: 0044710) and catalytic activity (GO: 0003824)
3.5% DNA transposons (Supplementary Table 8). Retro- (Supplementary Table 9). Furthermore, a phylogenetic
transposons are the main components of transposons, tree was constructed with 166 single-copy genes in
with Copia and Gypsy (37.92% and 29.06%, respectively) C. lavandulifolium, and the other 10 species showed
being the most common. Compared with other Aster- that C. lavandulifolium diverged from C. nankingense at
aceae species, LTR expansion of C. lavandulifolium was approximately 7.2 Mya (Figure 1b). We also compared
detected at ∼1.25 Mya, which was very close to that of gene family expansion and contraction among the 11
C. nankingense (∼1.45 Mya) (Figure 1c). Bursts of Copia species to examine the evolution of the C. lavandulifolium
and Gypsy occurred at ∼1.25 Mya, which was consis- genome (Figure 1b). The results showed that 1305 and
tent with the LTR expansion time of C. lavandulifolium 453 gene families were expanded and contracted in C.
(Figure 1d). This result indicated that LTR expansion in C. lavandulifolium, respectively (Figure 1b). The gene families
lavandulifolium was mainly driven by bursts of Copia and that were expanded in the C. lavandulifolium genome were
Gypsy. enriched in flower development-related GO terms and
4 | Horticulture Research, 2022, 9: uhab022 cell synthesis-related GO terms (Supplementary Table expressed genes between the two samples are shown 10). in Supplementary Fig. 9b. Genes with an adjusted P Whole-genome duplication (WGD) is one of the most value
Wen et al | 5
Downloaded from https://academic.oup.com/hr/article/doi/10.1093/hr/uhab022/6510191 by guest on 04 March 2022
Figure 2. Transcriptomic profiling analysis of six important stages of capitulum development in C. lavandulifolium. a, The morphology of six
developmental stages across samples. Stage 1 (S1), vegetative stage; stage 2 (S2), doming stage; stage 5 (S5), the initiation stage of disc f loret primordia;
stage 6 (S6), the initiation stage of ray f loret primordia, at which stage disc and ray f loret primordia began to differentiate; stage 9 (S9), the middle
stage of corolla primordia differentiation; and stage 10 (S10), the final stage of corolla primordia differentiation. FP, foliage primordia; Br, bract; DFP,
disc f loret primordia; and RFP, ray f loret primordia. b, Weighted gene coexpression network analysis of six developmental stages of f lowers and leaves
in C. lavandulifolium. c, Expression patterns of genes in gray modules that might be involved in the development of stage 6. d, Candidate hub genes
involved in the genetic regulatory networks of stage 6 (gray). Yellow triangles represent the cis-regulatory motifs of those genes, green circles represent
the gene ID number, and blue hexagons represent the GO terms that were enriched. e, Heatmap of NAM/CUC-LIKE and LOB30-LIKE at different
developmental stages of C. lavandulifolium.
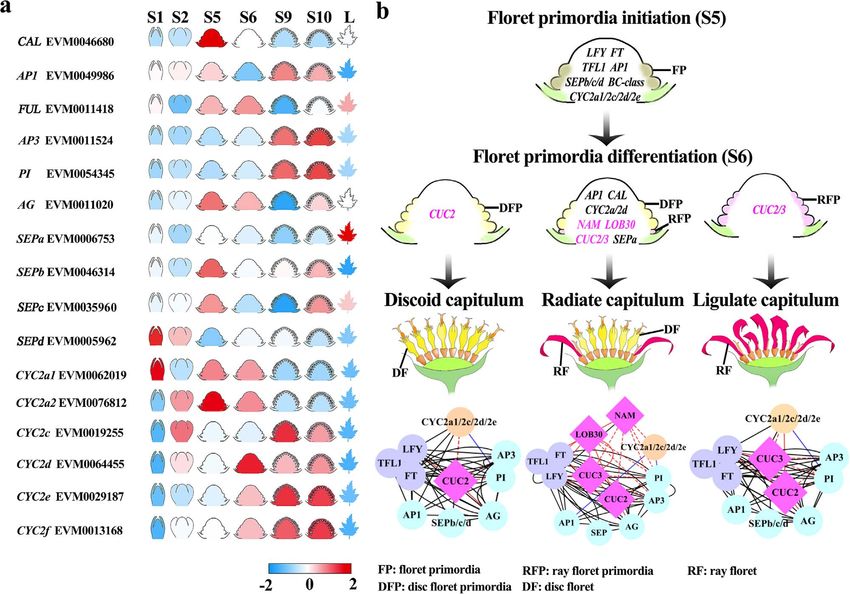
of them expanded during the evolutionary history of the only disc floret primordia began to initiate (Figure 4a and
C. lavandulifolium genome (Supplementary Figure 16 and Supplementary Figure 17). CAL was located on chr05, and
Supplementary Table 15). In our studies, B- and C-class SEPa was located on chr02 (Supplementary Table 14). The
genes were highly expressed when the second and third A-class gene FRUITFULL (FUL, EVM0011418) was mainly
whorls of disc and ray f lorets began to initiate (Figure 4a, expressed at stages 5 and 6, especially at stage 6, when
Supplementary Figures 17 and 19). We identified three A- ray floret primordia began to initiate on the capitulum
class genes and four E-class genes expressed during early (Figure 4a and Supplementary Figure 17). These results
capitulum development (Figure 4a, Supplementary Fig- indicated that the functions of CAL, SEPa and FUL might
ures 17 and 18). Notably, the A-class gene CAULIFLOWER regulate the differentiation of disc and ray florets.
(CAL, EVM0046680) and E-class gene SEPELLATAa (SEPa, CYC2-LIKE genes are known to regulate the symmetry
EVM0006753) were highly expressed at stage 5, when of flowers [26]. Homology analysis showed that there6 | Horticulture Research, 2022, 9: uhab022
Downloaded from https://academic.oup.com/hr/article/doi/10.1093/hr/uhab022/6510191 by guest on 04 March 2022
Figure 3. The expression patterns of NAM/CUC and LOB30 homologous genes in ten Asteraceae species. Notes: Transcriptomic profiling of
development stages in radiate, discoid and ligulate capitula. Stage 6 of the radiate capitulum has both disc and ray f loret primordia, stage 6 of the
discoid capitulum only has disc f loret primordia, and stage 6 of the ligulate capitulum only has ray f loret primordia. RF, ray f lorets; DF, disc f lorets; FP,
foliage primordia; Br, bract; DFP, disc f loret primordia; and RFP, ray f loret primordia.
Figure 4. The probable gene regulation mechanism in the development of different capitula types. a, The expression patterns of ABCE-class and
CYC2-LIKE genes in C. lavandulifolium during capitulum development. b, Gene and protein interactions involved in different capitula types. The dark
and red solid lines represent protein interactions predicted by STRING. The blue solid lines represent the protein interaction verified by experiments in
Wen et al. 2019. The red dotted lines represent protein interactions during capitulum development in Asteraceae.
were eight CYC2-LIKE genes in the C. lavandulifolium showed increasing expression with the development
genome; these genes were mainly distributed on chr06, process, except CYC2a1 (EVM0062019), which was mainly
chr07 and chr08 (Supplementary Figure 20, Supplemen- expressed at vegetative stage 1 (Figure 4a and Supple-
tary Tables 14 and 16). Most of the CYC2-like genes mentary Figure 21). Moreover, tandem duplication eventsWen et al | 7
in the C. lavandulifolium genome drove the duplication of using Nanopore and PacBio technologies with assis-
CYC2c/2d/2e/2f , and this gene set might have undergone tance from a Hi-C heatmap. The reference genome of
subfunctionalization during the evolution of the C. C. lavandulifolium that we obtained displayed higher
lavandulifolium genome (Supplementary Figure 22). CYC2c integrality and accuracy than that for C. nankingense at
and CYC2d showed different expression patterns, which the chromosome level3 . Compared with the assembled
indicated that they were subfunctionalized (Figure 4a genomes of other Asteraceae species, the scaffold N50 of
and Supplementary Figure 23). The expression patterns the C. lavandulifolium genome was the longest, at up to
of CYC2e and CYC2f were similar, which indicated that 300 Mb.
these two genes had redundant functions in regulating Flower crops with new and unique flower types could
capitulum development (Figure 4a and Supplementary have great economic value, and flower type modification
Figure 21). Notably, the duplication of CYC2a was unique is an important goal of ornamental breeding. Genetic
in the genus Chrysanthemum, and both copies were manipulation of floral development-related genes is
Downloaded from https://academic.oup.com/hr/article/doi/10.1093/hr/uhab022/6510191 by guest on 04 March 2022
expressed during the early capitulum development an effective method to directionally modify flower
stage, especially CYC2a2 (EVM0076812), which was highly types. Previous studies have found that modifying
expressed at stage 5 (Figure 4a, Supplementary Figures the expression of floral-related genes, such as MADS-
21 and 22). This result suggested that CYC2a2 might box and TCP transcription factors, could directionally
be a unique gene in regulating capitulum development improve the floral types [15]. These studies mainly
in the genus Chrysanthemum. In summary, five ABCE- concentrate on ABCE-class and CYC2-LIKE genes to
class and CYC2-LIKE genes were mainly expressed at influence the identity of floral organs and the symmetry
stages 5 and 6, which indicated that they might be of a single flower [25]. However, the flowers of some
involved in disc and ray f loret differentiation (Figure 4a). plants condense to form inflorescences, which is called
However, homologous genes of ABCE-class and CYC2- “pseudanthium”, that usually have higher ornamental
LIKE in other Asteraceae species did not show obvious value. The regulation of this complex inflorescence
expression differences among different capitula types remains to be clarified. Chrysanthemums, as one of the
(Supplementary Figure 23). most valuable ornamental plants, are famous for their
Protein interactions among these candidate genes diverse capitulum types. The modification of capitulum
were predicted by STRINGS and previous studies types in chrysanthemum will also provide insights for
(Figure 4b). In addition to ABCE-class and CYC2-LIKE other inflorescence type modifications. However, the
genes, inf lorescence meristem-related genes (FT, TFL1 molecular mechanism of capitulum development is
and LFY) are also involved in capitulum development. hindered by the complexity of flower types and the lack
However, the expression levels of FT, TFL1 and TFL at stage of genomic data.
5 and stage 6 were relatively lower than those at other The expression patterns of ABCE-class genes in the
developmental stages (Supplementary Figure 24). In the capitulum developmental process are different from
discoid capitulum, CUC2 interacted with LFY and AG to those in a single flower, but their functions are relatively
regulate the initiation of disc f loret primordia (Figure 4b). conserved and mainly play roles in regulating the
When CUC2 interacted with CUC3, the expression of identity of the four floral organ whorls during disc and
these two genes could promote the development of floret ray floret development [15]. The present study found
primordia into ray f lorets (Figure 4b). During radiate that most of the ABCE-class genes began to be highly
capitulum development, the existence of NAM and expressed during stage 9 - stage 10, the stages in which
LOB30 contributed to the differentiation of ray and disc the floral organs initiate on the disc and ray florets. The
floret primordia. LOB30 could interact with LFY, TFL1, CYC2-LIKE genes in Asteraceae expanded significantly.
CUC2, CUC3 and NAM, indicating its hub role in the CYC2-LIKE genes have been subfunctionalized and
genetic regulatory network (Figure 4b). Overall, NAM and neofunctionalized in regulating the differentiation of
LOB interacted with not only inf lorescence meristem- disc and ray florets [27]. Previous transgenic studies of
related genes (LFY) but also f loral organ identity genes CYC2c and CYC2d in C. lavandulifolium have changed the
and CYC2-LIKE genes during capitulum development, length of ray florets to some extent, which indicates that
indicating the hub roles of NAM and LOB30 in the CYC2-LIKE regulates floret development via the control of
differentiation of disc and ray f lorets on the radiate dorsal petal elongation [27, 28]. Combined with our wide
capitulum. survey of CYC2-LIKE genes during the key developmental
stage of C. lavandulifolium capitulum, CYC2e/2f may
be redundant with CYC2c/2d, and CYC2a may have
Discussion evolved a new function in the genus Chrysanthemum.
To date, the genomes of 16 Asteraceae species have Overall, the gene expression and evolution at the
been sequenced, including L. sativa, E. breviscapus, genome level showed that the ABCE-class and CYC2-LIKE
Artemisia annua, C. nankingense, C. seticuspe, H. annuus, genes contributed to the capitulum development of C.
and Mikania micrantha (Supplementary Table 1). In lavandulifolium.
this study, a high-quality chromosomal-scale reference However, transgenic studies of ABCE-class and
genome of C. lavandulifolium was successfully obtained CYC2-LIKE genes in Gerbera, Senecio vulgaris and C.8 | Horticulture Research, 2022, 9: uhab022
lavandulifolium did not alter the identity of disc and ray nk-mer represents the k-mer total number, and daverage
florets, although some of these genes could change k-mer is the average k-mer depth). Genome sequencing
the morphology of the two kinds of f lorets or cause was performed using SMRT sequencing on a PacBio RS
the complete loss of the inf lorescence meristem15 . II sequencer (Pacific Biosciences, Menlo Park, CA, USA)
It appears that ABCE-class and CYC2-like genes in following the manufacturer’s standard protocol, and
Asteraceae are not the hub genes in the regulatory 201.85 Gb PacBio data using SMRT analysis software
network of disc and ray f loret differentiation. WGCNA v1.2 [34] were acquired. To further improve the genomic
showed that the hub genes regulating disc and ray assembly quality, 193.88 Gb of clean data were generated
floret differentiation in the capitulum were NAM and using a PromethION sequencer (Pacific Biosciences,
LOB30, and NAM/CUC and LOB30 subfamily genes not Menlo Park, CA, USA).
only regulate the initiation and orientation of organ
primordia in early f lower development but also respond Genome assembly and quality assessment
Downloaded from https://academic.oup.com/hr/article/doi/10.1093/hr/uhab022/6510191 by guest on 04 March 2022
to inf lorescence meristem-related genes and floral For the PacBio RSII platform data, longer subreads were
identity genes [29–31]. Based on the hub roles of NAM selected by the error correction module of canu v1.5
and LOB30 in stages 5 and 6, we concluded that the [35]. Raw overlapping subreads were detected through
two genes might regulate the differentiation of disc and the highly sensitive overlap detection program MHAP
ray f loret primordia in early capitulum development v2.1 [36], and the error correction of these data was
and that they could interact with downstream genes carried out by the Falcon sense method v0.40 (“correct-
to regulate the f loral organ identity and development of edErrorRate = 0.025”) [37]. The error-corrected subreads
each f loret on the capitulum. Duplication of NAM and its were used to generate a draft assembly in WTDBG v2.5
chromosome position in the C. lavandulifolium genome (https://github.com/ruanjue/wtdbg). Iterative polishing
indicated that NAM might be a special key gene in by Pilon v1.22 [38] was achieved by aligning adapter-
regulating the diverse capitula types of chrysanthemum. trimmed and paired-end Illumina reads to the PacBio
Protein interactions showed that these hub genes could draft genome. Clean nanopore data were acquired via
interact with LFY, which has been proven to regulate sequencing on the PromethION platform, and these data
ray floret development in Gerbera [31]. Previous studies were corrected with the same method described above.
supported the idea that the capitulum was derived from The draft genome assembled by WTDBG v2.5 (https://
a cyme in which peripheral branches were inhibited github.com/ruanjue/wtdbg) was corrected three times
[32]. The interaction of NAM/CUC and LOB30 regulated by Racon v1.3.3 [39] by aligning adapter-trimmed and
the expression of LFY to prevent the development of paired-end Illumina reads to the Nanopore draft genome.
peripheral branches and promote peripheral ray floret Then, the PacBio draft genome as a query input was
primordia in different capitula types. In conclusion, aligned against the Nanopore draft genome using MUM-
the C. lavandulifolium genome presented in this study mer v4.0.0 [40]. The PacBio draft genome and Nanopore
provides a powerful reference for the further assembly draft genome were then merged using quickmerge v0.3.0
of complex genomes, especially for the deciphering [21]. This merged draft genome was polished by Racon
of chrysanthemum genomes. Based on comparative v1.3.3 [39] and Pilon v1.22 [38]. The mapping depth was
genomic and transcriptomic analyses, we identified hub obtained by aligning corrected Nanopore sequencing
genes that might be involved in the identity of disc and data to the merged assembly by minimap2 v2.17 [41]
ray f lorets, which could serve as candidate genes for with default parameters. Then, purge haplotigs v1.0.4
further genome editing to modify the capitula types in [22] was used to eliminate redundancy according to the
chrysanthemum. coverage depth and obtain the purged haplotig genome.
Ultimately, a C. lavandulifolium genome with a total
length of 3.10 Gb was obtained. The second-generation
Materials and methods sequencing data, core gene completeness and BUSCOs
Plant materials and sequencing were evaluated to verify the accuracy of the genome
The C. lavandulifolium G1 line was collected and cultured assembly.
at Beijing Forestry University for genomic sequencing
[33]. The genomic DNA of the C. lavandulifolium G1 line Hi-C sequencing and assistant assembly
was extracted using a standard CTAB protocol. The Hi-C is a technology derived from chromosome
paired-end libraries were sequenced on the Illumina conformation capture technology that utilizes high-
HiSeqTM 4000 sequencing platform (Illumina, San Diego, throughput sequencing data and is mainly used to assist
CA, USA). The 27-mer frequencies were generated using in genome assembly. We constructed Hi-C fragment
135.61 Gb of high-quality PE reads (51.43 ×), and a total libraries with insert sizes of 300–700 bp, as illustrated
of 1 × e11 k-mers were obtained by a customized Perl in Rao et al. [42], and sequenced them using the Illumina
script. The main peak value representing the average platform [42]. Before chromosome assembly, we first
k-mer depth was 38. A modified formula was used performed a preassembly for the error correction of
to estimate the C. lavandulifolium genome size, G = scaffolds, which required splitting the scaffolds into
nk-mer/daverage k-mer (G represents the genome size, segments of 50 kb, on average. Then, the Hi-C dataWen et al | 9
were mapped to these segments using BWA aligner GeneMarkS-T (version 5.1) [56] and TransDecoder v2.0
v0.7.10-r789 [43]. We retained the uniquely mapped (http://transdecoder.github.io). Finally, EVM v1.1.1 [57]
data to assemble the genome using LACHESIS [44] with was used to integrate the prediction results, with
the following parameters: CLUSTER_MIN_RE_SITES = 80; the following parameters: —min_intron_length 2 —
CLUSTER_MAX_LINK_DENSITY = 2; terminal_intergenic_re_search 10 000. MicroRNA and
CLUSTER_NONINFORMATIVE_RATIO = 2; rRNA were identified by BLAST with 1e-10 based on
ORDER_MIN_N_RES_IN_TRUN = 16; and the Rfam database [58]. Transfer RNAs (tRNAs) were
ORDER_MIN_N_RES_IN_SHREDS = 16. To further address predicted using tRNAscan-SE [59]. Repetitive sequences
the redundant sequences, we manually checked any were predicted using Repeat Masker [60].
two segments that showed inconsistent connections
with the raw scaffold. The detailed workf low schema
for the assembly pipeline of the chromosome-scale Gene family identification, genome evolution
Downloaded from https://academic.oup.com/hr/article/doi/10.1093/hr/uhab022/6510191 by guest on 04 March 2022
C. lavandulifolium genome is shown in Supplementary analysis and species tree construction
Figure 24. The alignments of protein sequences were performed
using Diamond v0.9.29.130 (http://www.diamondsea
Transcriptome sequencing rch.org/index.php) [61] with an E-value of 0.001. Orthol-
The reproductive-stage leaf and developmental series ogous and paralogous gene families were identified
of capitula (stage 1, stage 2, stage 5, stage 6, stage by OrthoFinder v2.3.7 [62] with default parameters. A
9 and stage 10) of the C. lavandulifolium G1 line were phylogenetic tree based on the concatenated sequence
sampled to establish transcriptomic profiling (Figure 2a, alignment of 166 single-copy gene families from C. lavan-
Supplementary Table 12). The reproductive-stage leaves, dulifolium and 10 other plant species was constructed
vegetative buds and reproductive buds of nine other using IQ-TREE with the selected optimal sequence
species of Asteraceae were also sampled to construct evolution model (−m JTT + F + R5) and with ultrafast
libraries for RNA-seq (Figure 2b, Supplementary Table 13 bootstrapping [63]. MCMCTREE of PAML (v4.9) was used
and Supplementary Figure 15). Total RNA was extracted to estimate the divergence times [64]. Ks-based age dis-
using a Plant RNA Rapid Extraction Kit (HUAYUEYANG tributions were analyzed by using PAML to calculate the
Biotechnology, Beijing, China) and treated with RNase- synonymous mutation rate (Ks) values. LTR sequences
free DNase I to digest the DNA. After assessing the purity were identified and filtered using LTR_FINDER v1.07
and integrity of RNA using the Agilent 2100 Bioanalyzer (score = 6) [65]. Then, the flanking sequences of the LTRs
(Agilent Technologies, Palo Alto, Calif.) and the ABI were extracted and compared using MAFFT (parameters:
StepOnePlus Real-Time PCR System (Applied Biosystems, —localpair —maxiterate 1000) [66]. The distance K was
Waltham, MA, USA), the constructed libraries were calculated by the Kimura model using EMBOSS v6.6.0
sequenced on an Illumina HiSeqTM 2500 sequencing plat- [67]. The formula for calculating time is T = K/(2 × r) with
form (Illumina, San Diego, CA, UAS) [45]. The clean reads the molecular clock r = 7 × 10−9 mutations per site per
were aligned to our de novo genome of C. lavandulifolium year.
using TopHat2 (version 2.0.7) [46] and then assembled
using Cuff links [47] after removing the connectors of the
low-quality sequences and raw reads. The protein-coding WGCNA of flower development in C.
genes were annotated against the NCBI NR (http://www. lavandulifolium
ncbi.nlm.nih.gov), SwissProt, GO, COG, KOG, eggNOG, and To analyze genes involved in the six capitulum develop-
KEGG databases. Gene expression was calculated using mental stages of C. lavandulifolium, weighted correlation
Cuffquan and CuffnormGene in Cuff links [47]. network analysis (WGCNA) was performed using the R
package [68]. The soft thresholding power was set to 7
Gene annotation to construct an adjacency matrix of genes with different
To better predict the protein-coding genes, a pipeline that expression patterns, and the topological overlap matrix
combined de novo gene prediction, unigene prediction (TOM) similarity algorithm was used to transform the
and homologous species prediction was used. For de novo adjacency matrix into a topological overlap matrix to
prediction, Genscan [48], Augustus v2.4 [49], Glimmer reduce noise and false correlations. Then, all DEGs were
HMM v3.0.4 [50], GeneID v1.4 [51], and SNAP [52] were hierarchically clustered based on TOM similarity. Hier-
used; for homology prediction, GeneWise v2.2.0 [53] was archical clustering was performed by Dynamic Hybrid
used with C. lavandulifolium protein sequences, and a Tree Cut [69]. The genes in different colored modules
minimum of 50% coverage was set to the determined were converted to module eigengenes using the first
value using gene models. For unigene prediction, principal component. The different capitulum develop-
Illumina reads were filtered to remove adaptors and ment stages in C. lavandulifolium were also correlated with
trimmed to remove low-quality bases. Processed reads the eigengenes of each module to find the key mod-
were aligned to the reference genome, and then the ule associated with capitulum development. The expres-
transcripts were assembled using Hisatv2.0.4 [54] and sion heatmap of candidate genes was constructed using
Stringtie [55]. Coding sequences were predicted using TBtools [70].10 | Horticulture Research, 2022, 9: uhab022
Evolution and expression analysis of key chrysanthemum (chrysanthemum × morifolium Ramat.). Horticul-
candidate gene families ture Research. 2020;7:108.
The evolution and expression of genes related to capitu- 2. He SM, Dong X, Zhang G et al. High quality genome of Erigeron
breviscapus provides a reference for herbal plants in Asteraceae.
lum development in C. lavandulifolium were investigated.
Mol Ecol Resour. 2020;00:1–17.
Based on the previously described results for transcrip-
3. Song C, Liu Y, Song A et al. The chrysanthemum nankingense
tome analysis, we chose key gene families for further
genome provides insights into the evolution and diversifica-
analysis. The protein sequences of those candidate gene tion of chrysanthemum flowers and medicinal traits. Mol Plant.
families were scanned using BLASTP and HMMER. For 2018;11:1482–91.
BLASTP and HMMER, initial gene sets were filtered with 4. Hirakawa H, Sumitomo K, Hisamatsu T et al. De novo whole-
a default cutoff E-value of 1e-5. Then, phylogenetic trees genome assembly in chrysanthemum seticuspe, a model species
were established using FastTree (v2.1) [71] and modified of chrysanthemums, and its application to genetic and gene
by ITOL (https://itol.embl.de/). discovery analysis. DNA Res. 2019;26:195–203.
Downloaded from https://academic.oup.com/hr/article/doi/10.1093/hr/uhab022/6510191 by guest on 04 March 2022
5. Wellmer F, Riechmann JL. Gene networks controlling the initia-
tion of flower development. Trends Genet. 2010;26:519–527.
Acknowledgments 6. Thomson B, Wellmer F. Molecular regulation of flower develop-
This work was supported by grants from the National ment. Curr Top Dev Biol. 2019;131:185–210.
7. Krizek BA, Fletcher JC. Molecular mechanisms of flower devel-
Natural Science Foundation of China (No. 31530064)
opment: an armchair guide. Nature Rev Genet. 2005;6:688–98.
and National Key Research and Development Project
8. Chen F, Song Y, Li X et al. Genome sequences of horticul-
(2018YFD1000403). We are particularly thankful to Xia tural plants: past, present, and future. Horticulture Research.
Xu and Guanghui Zhang for providing T. kok-saghyz and 2019;6:112.
E. breviscapus. We are also grateful to Hongqing Ling and 9. Krizek BA. eLS. London: Wiley; 2020.
Yalong Guo for their helpful suggestions on our work. 10. Zhang QG, Liu KW, Li Z et al. The Apostasia genome and the
evolution of orchids. Nature. 2017;549:379–83.
11. Li MM, Zhang D, Gao Q et al. Genome structure and evolution of
Author contributions Antirrhinum majus L. Nature Plants. 2019;5:174–83.
S.L.D., L.S.Z., Y.N.J., and X.H.W. conceived and designed 12. Zhang LS, Chen F, Zhang X et al. The water lily genome and the
the study. S.L.D., L.S.Z., and J.Y.N. discussed and modi- early evolution of flowering plants. Nature. 2020;577:79–84.
fied the study results. X.H.W., J.Z.L., Q.G., P.X., C.F.L, Y.P., 13. Ding L, Song A, Zhang X et al. The core regulatory networks
and hub genes regulating flower development in Chrysanthemum
H.L., Q.L.Z., X.Y.W., C.F.M., and K.G. prepared the materi-
morifolium. Plant Mol Biol. 2020;103:669–88.
als, conducted the experiments, analyzed the data and
14. Elomaa P, Zhao Y, Zhang T et al. Flower heads in Asteraceae -
prepared the results. X.H.W., J.Z.L., and L.C.F. wrote the recruitment of conserved developmental regulators to control
manuscript. K.D.R. and Y.H. were involved in data inter- the flower-like inflorescence architecture. Horticulture research.
pretation and finalizing the manuscript draft. All authors 2018;5:1–10.
read and approved the final draft. 15. Zoulias N, Duttke SHC, Garcês H et al. Auxin and pattern for-
mation of the Asteraceae flower head (capitulum). Plant Physiol.
2019;179:391–401.
Data availability 16. Li F, Lan W, Zhou Q et al. Reduced expression of CbUFO is
The raw sequence data and assembly of C. lavandulifolium associated with the phenotype of a flower-defective Cosmos
genome sequencing and RNA sequencing have been bipinnatus. Int J Mol Sci. 2019;20:2503.
17. Dai SL, Zhang CJ, Chen J et al. Advances of researches on phy-
deposited in NCBI (PRJNA681093). The final assembly and
logeny of Dendranthema and origin of chrysanthemum. Journal of
annotation of the C. lavandulifolium genome are available
Beijing Forestry University. 2002;24:234–8.
at GenBank under accession number JAHFWF000000000.
18. Yang LW, Wen XH, Fu JX et al. ClCRY2 facilitates floral transition
The accession numbers of other transcriptome data in chrysanthemum lavandulifolium by affecting the transcription
sequenced in the present study are shown in Supple- of circadian clock-related genes under short-day photoperiods.
mentary Table 13. Horticulture Research. 2018;5:58.
19. Wen XH, Qi S, Huang H et al. The expression and interactions of
Competing interests ABCE-class and CYC2-like genes in the capitulum development of
chrysanthemum lavandulifolium and C. × morifolium. Plant Growth
The authors declare no competing interests. Regul. 2019;88:205.
20. Qi S, Yang L, Wen X et al. Reference gene selection for RT-
Supplementary data qPCR analysis of flower development in Chrysanthemum mori-
folium and chrysanthemum lavandulifolium. Front Plant Sci. 2016;7:
Supplementary data is available at Horticulture Research 651.
Journal online. 21. Chakraborty M, Baldwin-Brown JG, Long AD et al. Contiguous
and accurate de novo assembly of metazoan genomes with
modest long read coverage. Nucleic Acids Res. 2016;44:e147.
References
22. Roach MJ, Schmidt SA, Borneman AR. Purge Haplotigs: allelic
1. Song XB, Xu Y, Gao K et al. High-density genetic map construc- contig reassignment for third-gen diploid genome assemblies.
tion and identification of loci controlling flower-type traits in BMC Bioinformatics. 2018;19:460.Wen et al | 11
23. Qiao X, Li Q, Yin H et al. Gene duplication and evolution in recur- 44. Burton JN, Adey A, Patwardhan RP et al. Chromosome-scale
ring polyploidization - diploidization cycles in plants. Genome scaffolding of de novo genome assemblies based on chromatin
Biol. 2019;20:38. interactions. Nat Biotechnol. 2013;31:1119–25.
24. Badouin H, Gouzy J, Grassa CJ et al. The sunflower genome 45. Singh KS, Wu Y, Ghosh JS et al. OPEN RNA-sequencing reveals
provides insights into oil metabolism, flowering and Asterid global transcriptomic changes in Nicotiana tabacum responding
evolution. Nature. 2017;546:7675. to topping and treatment of axillary-shoot control chemicals.
25. Liu B, Yan J, Li W et al. Mikania micrantha genome provides Sci Rep. 2016;5:18148.
insights into the molecular mechanism of rapid growth. Nature. 46. Kim D, Pertea G, Trapnell C et al. TopHat2: accurate alignment of
Communications. 2020;11:1. transcriptomes in the presence of insertions, deletions and gene
26. Spencer V, Kim M. Re"CYC"ling molecular regulators in the fusions. Genome Biol. 2013;14:4.
evolution and development of flower symmetry. Semin Cell Dev 47. Trapnell C, Roberts A, Goff L et al. Differential gene and transcript
Biol. 2018;79:16–26. expression analysis of RNA-seq experiments with TopHat and
27. Chen J, Shen CZ, Guo YP et al. Patterning the Asteraceae Capit- cufflinks. Nat Protoc. 2012;7:562–78.
Downloaded from https://academic.oup.com/hr/article/doi/10.1093/hr/uhab022/6510191 by guest on 04 March 2022
ulum: duplications and differential expression of the flower 48. Burge C, Karlin S. Prediction of complete gene structures in
symmetry CYC2-like genes. Frontiers. Plant Sci. 2018;9:1–14. human genomic DNA. J Mol Biol. 1997;268:78–94.
28. Huang CH, Zhang C, Liu M et al. Multiple polyploidization events 49. Stanke M, Waack S. Gene prediction with a hidden Markov
across Asteraceae with two nested events in the early his- model and a new intron submodel. Bioinformatics. 2003;19:
tory revealed by nuclear phylogenomics. Mol Biol Evol. 2016;33: ii215–25.
2820–35. 50. Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM:
29. Zadnikova P, Simon R. How boundaries control plant develop- two open source ab initio eukaryotic gene-finders. Bioinformatics.
ment. Curr Opin Plant Biol. 2014;17:116–25. 2004;20:2878–9.
30. Mara C, Manrique S, Cuesta C et al. CUP-SHAPED COTYLEDON1 51. Blanco E, Genís P, Roderic G. Using GENEID to identify genes. Curr
(CUC1) and CUC2 regulate cytokinin homeostasis to determine Protoc Bioinformatics. 2007;18:1.
ovule number in Arabidopsis. J Exp Bot. 2018;69:5169–76. 52. Korf I. Gene finding in novel genomes. BMC bioinformatics.
31. Rebocho AB, Kennaway JR, Bangham JA et al. Formation and 2004;5:59.
shaping of the antirrhinum flower through modulation of the 53. Birney E, Clamp M, Durbin R. GeneWise and Genomewise.
CUP boundary gene. Curr Biol. 2017;27:2610–2622.e3. Genome Res. 2004;14:988–95.
32. Zhao Y, Zhang T, Broholm SK et al. Evolutionary co-option 54. Kim D, Langmead B, Salzberg SL. HISAT: a fast-spliced aligner
of floral meristem identity genes for patterning of the with low memory requirements. Nat Methods. 2015;12:357–60.
flower-like Asteraceae inflorescence. Plant Physiol. 2016;172: 55. Pertea M, Pertea GM, Antonescu CM et al. StringTie enables
284–96. improved reconstruction of a transcriptome from RNA-seq
33. Wen XH, Pu Y, Liu Y. Effects of N, P and K nutrients on the growth reads. Bio/technology (Nature Publishing Company). 2015;33:
and development of chrysanthemum lavandulifolium based on 290–5.
BBCH scale. Advanced in Ornamental Horticulture of China. 2019;1: 56. Tang SYY, Alexandre L, Mark B. Identification of protein cod-
76–84. ing regions in RNA transcripts. Nucleic Acids Symp Ser. 2015;43:
34. Ramsköld D, Luo S, Wang YC et al. Full-length mRNA-Seq from e78.
single-cell levels of RNA and individual circulating tumor cells. 57. Haas BJ, Salzberg SL, Zhu W et al. Automated eukaryotic gene
Nat Biotechnol. 2012;30:777–82. structure annotation using EVidenceModeler and the program
35. Koren S, Walenz BP, Berlin K et al. Canu: scalable and accurate to assemble spliced alignments. Genome Biol. 2008;9:R7.
long-read assembly via adaptive k-mer weighting and repeat 58. Griffiths-Jones S, Moxon S, Marshall M et al. Rfam: annotat-
separation. Genome Res. 2017;27:722–36. ing non-coding RNAs in complete genomes. Nucleic Acids Res.
36. Drake JP, Berlin K, Koren S et al. Assembling large genomes with 2004;33:D121–4.
single-molecule sequencing and locality-sensitive hashing. Nat 59. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detec-
Biotechnol. 2015;33:623–30. tion of transfer RNA genes in genomic sequence. Nucleic Acids
37. Chin CS, Peluso P, Sedlazeck FJ et al. Phased diploid genome Res. 1997;25:955–64.
assembly with single-molecule real-time sequencing. Nat Meth- 60. Tarailo-Graovac M, Chen N. Using RepeatMasker to identify
ods. 2016;13:1050–4. repetitive elements in genomic sequences. Curr Protoc Bioinfor-
38. Walker BJ, Abeel T, Shea T et al. Pilon: an integrated tool for com- matics. 2009;25:1.
prehensive microbial variant detection and genome assembly 61. Buchfink B, Xie C, Huson DH. Fast and sensitive protein align-
improvement. PLoS One. 2014;9:e112963. ment using diamond. Nat Methods. 2015;12:59–60.
39. Vaser R, Sović I, Nagarajan N et al. Fast and accurate de novo 62. Emms DM, Kelly S. OrthoFinder: solving fundamental biases in
genome assembly from long uncorrected reads. Genome Res. whole genome comparisons dramatically improves orthogroup
2017;27:737–46. inference accuracy. Genome Biol. 2015;16:157.
40. Marcais G, Delcher AL, Phillippy AM et al. MUMmer4: a fast 63. Lam-Tung N, Schmidt HA, von Haeseler A et al. IQ-TREE: a fast
and versatile genome alignment system. PLoS Comput Biol. and effective stochastic algorithm for estimating maximum-
2018;14:e1005944. likelihood phylogenies. Molecular Biology & Evolution. 2015;1:
41. Li H. Minimap2: pairwise alignment for nucleotide sequences. 268–74.
Bioinformatics. 2018;34:3094–100. 64. Rannala YB. Bayesian estimation of species divergence times
42. Rao SS, Huntley MH, Durand NC et al. A 3D map of the human under a molecular clock using multiple fossil calibrations with
genome at kilobase resolution reveals principles of chromatin soft bounds. Mol Biol Evol. 2006;23:212–26.
looping. Cell. 2014;159:1665–80. 65. Xu Z, Wang H. LTR_FINDER: an efficient tool for the pre-
43. Li H, Durbin R. Fast and accurate long-read alignment with diction of full-length LTR retrotransposons. Nucleic Acids Res.
burrows-wheeler transform. Bioinformatics. 2010;26:589–95. 2007;35:W265–8.12 | Horticulture Research, 2022, 9: uhab022
66. Katoh K, Standley DM. MAFFT multiple sequence alignment 69. Langfelder P, Zhang B, Horvath S et al. Defining clusters from
software version 7: improvements in performance and usability. a hierarchical cluster tree: the dynamic tree cut package for R.
Molec Biol Evol. 2013;30:772–80. Bioinformatics. 2008;24:719–20.
67. Rice P, Longden I, Bleasby A. EMBOSS: the European 70. Chen C, Chen H, Zhang Y et al. TBtools: an integrative toolkit
molecular biology open software suite. Trends Genet. 2000;16: developed for interactive analyses of big biological data. Mol
276–7. Plant. 2020;13:1194.
68. Langfelder P, Horvath S. WGCNA: an R package for 71. Morgan NP, Dehal PS, Arkin AP et al. FastTree: computing large
weighted correlation network analysis. Bioinformatics. 2008;9: minimum evolution trees with profiles instead of a distance
559. matrix. Mol Biol Evol. 2009;7:1641–50.
Downloaded from https://academic.oup.com/hr/article/doi/10.1093/hr/uhab022/6510191 by guest on 04 March 2022You can also read

















































