J-aggregation enhanced thermally activated delayed fluorescence for amplified spontaneous emission
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
ll
OPEN ACCESS
Article
J-aggregation enhanced thermally activated
delayed fluorescence for amplified
spontaneous emission
Jinlong Zhu, Qing Liao, Han
Huang, Liyuan Fu, Meihui Liu,
Chunling Gu, Hongbing Fu
liaoqing@cnu.edu.cn (Q.L.)
hbfu@cnu.edu.cn (H.F.)
Highlights
Two organic crystalline
polymorphs are prepared by a
controlled self-assembly method
The excited-state dynamics are
controlled by aggregate effects
Fluorescence and thermally
activated delayed fluorescence
adjustment
Amplified spontaneous emission
is modulated by polymorphs
By controlling excited-state dynamics through aggregate effects, Zhu et al.
achieve the regulation of fluorescence and thermally activated delayed
fluorescence (TADF) amplified spontaneous emission (ASE) in organic crystalline
polymorphs.
Zhu et al., Cell Reports Physical Science 3,
100686
January 19, 2022 ª 2021
https://doi.org/10.1016/j.xcrp.2021.100686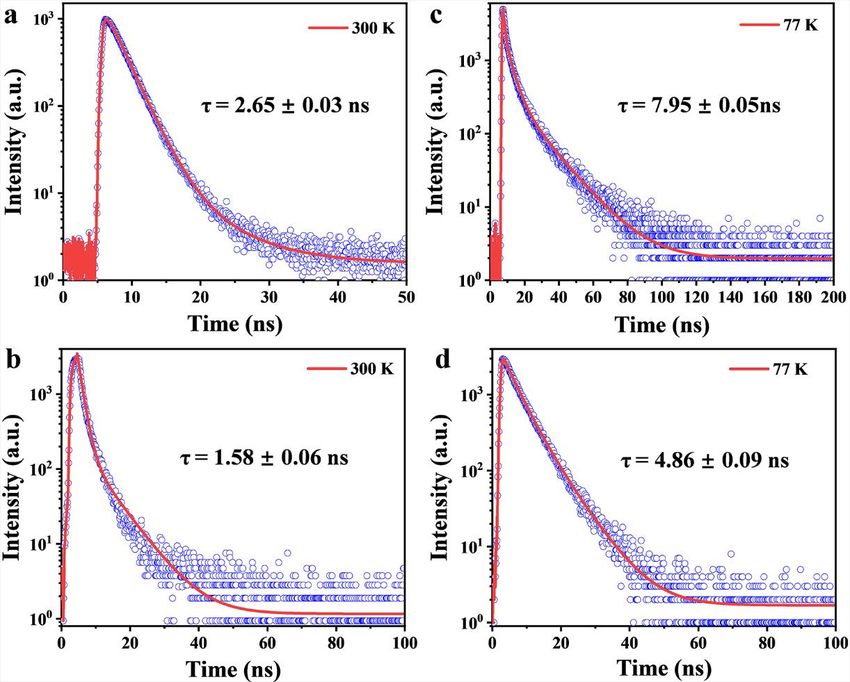
ll
OPEN ACCESS
Article
J-aggregation enhanced thermally activated delayed
fluorescence for amplified spontaneous emission
Jinlong Zhu,1 Qing Liao,1,* Han Huang,2 Liyuan Fu,1 Meihui Liu,1 Chunling Gu,3 and Hongbing Fu1,4,*
SUMMARY
Modulation of excited-state energy-level structures through
controlled molecular stacking arrangement provides an effective
strategy for tuning fluorescence and thermally activated delayed
fluorescence (TADF) amplified spontaneous emission (ASE) but
remains a critical challenge. Herein, we report the regulation of fluo-
rescence and TADF ASE in organic crystalline polymorphs by con-
trolling the excited-state dynamics through aggregate effects.
Experimental and theoretical studies show that green crystals
emit green fluorescence ASE, while red crystals emit red TADF
ASE because a higher degree of J-aggregation in red crystals signif-
icantly results in a substantial decrease of energy gaps between
singlet and triplet to 0.24 eV for the realization of a reverse inter-
system crossing process. Our results suggest that molecular packing
presents a powerful approach to tailor radiative channels that is
fundamentally important for tuning fluorescence and TADF ASE in
pure organic crystals.
INTRODUCTION
Organic solid-state lasers (OSSLs) as miniaturized coherent light sources have at-
tracted intense attention because of their potential for a wide range of applications
in optical communications, spectroscopy, high-throughput sensing, and three-
dimensional displays.1–5 Amplified spontaneous emission (ASE) is an essential
process similar to laser emission that allows the characterization of organic gain ma-
terials independent of an optical feedback structure.6 Generally, the abundant en-
ergy levels of organic materials facilitate the establishment of a quasi-four-level laser
system suitable for population inversion for ASE and lasing but also bring about a
number of optical and excitonic losses caused by triplet absorption and singlet-
triplet annihilation at high triplet exciton density.7 One approach for preventing
triplet accumulation is the use of triplet quenchers to scavenge the excess triplet ex-
citons,8,9 but in many cases this leads to uselessness of triplet excitons. More signif-
1BeijingKey Laboratory for Optical Materials and
icantly, reduction of triplet accumulation is extremely important for the development Photonic Devices, Department of Chemistry,
of electrically pumped OSSLs because 75% of the formed excitons are non-radiative Capital Normal University, Beijing 100048, China
triplets and only 25% are radiative singlets upon electrical excitation limited by spin 2TianjinKey Laboratory of Molecular
statistics.10,11 Such high-density triplets not only result in high lasing threshold and Optoelectronic Science, School of Chemical
Engineering and Technology, Tianjin University
poor stability of the devices but also severely limit the internal quantum efficiencies and Collaborative Innovation Center of Chemical
of electrically pumped OSSLs. Science and Engineering (Tianjin), Tianjin 300072,
P.R. China
3Institute
of Process Engineering, Chinese
Organic molecules capable of thermally active delayed fluorescence (TADF) may Academy of Sciences, Beijing 100190, China
provide an alternative strategy to use triplet excitons through an effective reverse 4Lead contact
intersystem crossing (RISC) process from a non-radiative triplet state (T1) to a radia- *Correspondence: liaoqing@cnu.edu.cn (Q.L.),
tive singlet state (S1) at room temperature.12,13 The RISC process would not only hbfu@cnu.edu.cn (H.F.)
diminish triplet accumulation but also promote up-conversion from triplets to https://doi.org/10.1016/j.xcrp.2021.100686
Cell Reports Physical Science 3, 100686, January 19, 2022 ª 2021 1
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).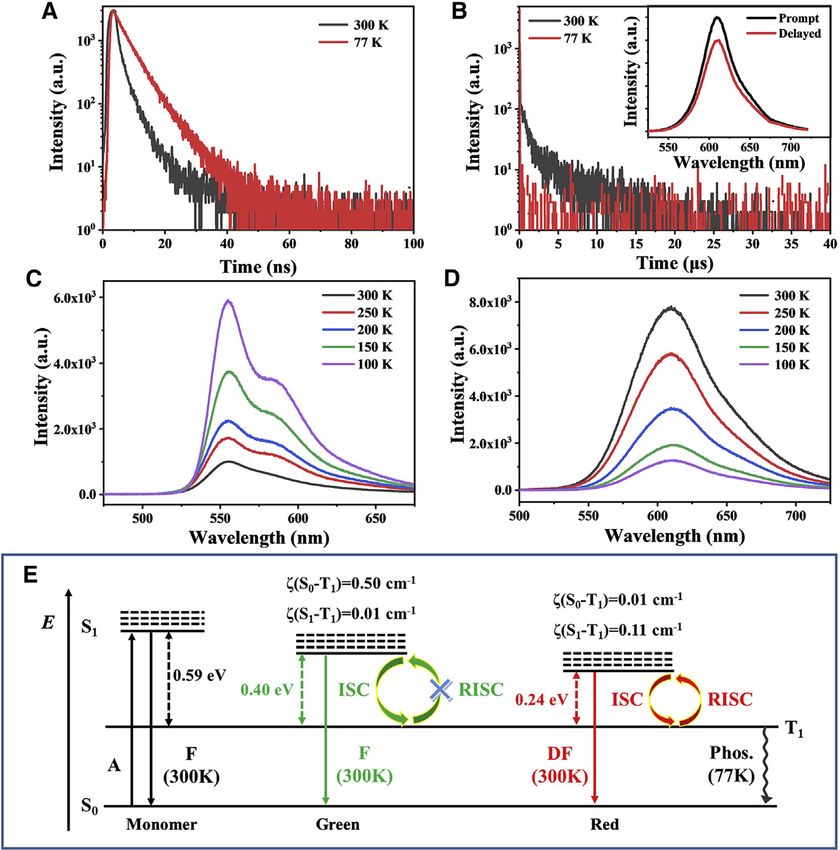
ll
OPEN ACCESS
Article
regenerate singlets to benefit ASE and lasing emission.14,15 In recent years, ASE and
lasing on the basis of TADF mechanisms have been successively demonstrated in
host-guest thin films doped with different TADF molecules14,16,17 and pure TADF
crystals.15 In contrast to the thin-film counterparts, organic nanostructures with high-
ly ordered molecular packing show superior optical and electronic properties
because of their absence of grain boundaries and low density of defects.14 It is worth
noting that ordered molecular packing into aggregates will endow materials new
energy-level structures and controllable excited-state processes. For example, ac-
cording to the Kasha exciton model,18 the splitting of pristine singlet excitons in
J-aggregates can generate lower energy exciton states with larger transition dipole
moments, while pristine triplet excitons are almost unchanged because of the signif-
icant difference in their transition dipole moments.19–21 Importantly, the reduction of
energy gaps between S1 and T1 (DEST) is expected to realize efficient TADF in prin-
ciple. Therefore, the control of the molecular packing (generally called polymorph)
would provide an alternative approach for exploring excited-state dynamics22 and
tailoring the radiative channels such as fluorescence and TADF emission, especially
avoiding the difficulty of time-consuming molecular synthesis. Note that transition
dipole moment is directly related to the stimulated emission cross section, an
important parameter in a laser gain medium. In practice, J-aggregates with larger
transition dipole moments bring out the faster radiative rate and facilitate the
improvement of gain coefficients for ASE processes. However, controlling of fluores-
cence and TADF ASE by aggregate-state effects and their intrinsic relationships
remain largely unexplored.
Here, aiming to realize the transition from fluorescent to TADF ASE in organic poly-
morphs, we design and synthesize a novel laser-gain molecule with a strong intramo-
lecular charge transfer (CT)15,17 on the basis of the strong electron-drawing acceptor
difluoroboron skeleton and a strong electron-donating donor indole moiety.
Through a facile solution self-assembly method, we successfully fabricate its green
and red microcrystals with different molecular stacking arrangements. Although
both belong to J-aggregations, the green and red crystals exhibit different energy
splitting of pristine singlet excitons, thereby resulting in a substantial decrease of
DEST from 0.40 to 0.24 eV. Experimental and theoretical studies show that green
crystals emit green fluorescence ASE, while red crystals emit red TADF ASE, because
a higher degree of J-aggregation in red crystals significantly reduces DEST for the
realization of the RISC process. Consequently, the larger radiative rate and triplet-
assisted ASE in red crystals means that their ASE threshold is nearly 4 times lower
than that of green crystals. Our results demonstrate that the exploration of molecular
packing might control fluorescence and TADF ASE and provide guidance for high-
performance electrically driven OSSLs.
RESULTS AND DISCUSSION
Synthesis and solution controllable self-assembly of two polymorphs
The organic difluoroboron derivative (E)-5-(ethoxycarbonyl)-2,2-difluoro-6-methyl-
4-(2-[1-methyl-1H-indol-2-yl]vinyl)-2H-1,3,2-dioxaborinin-1-ium-2-uide (Figure 1A)
was synthesized. Details on the synthesis and purification and characterization of
chemical properties of this compound are provided in the Supplemental information
(Note S1; Figure S1).
The polymorphs of this compound were prepared using a facile solution self-assem-
bly method. Briefly, 2 mL n-hexane was rapidly added to 200 mL of the stock
dichloromethane (CH2Cl2) solution (5 or 10 mg/mL) of the synthesized target com-
pound (Figure 1A). From fluorescence microscopy images, samples with bright
2 Cell Reports Physical Science 3, 100686, January 19, 2022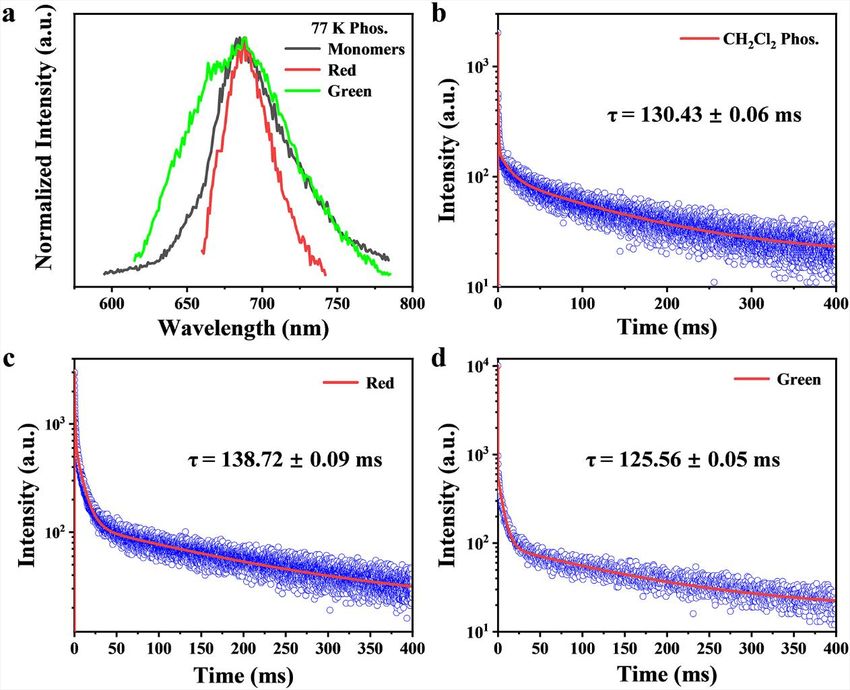
ll
OPEN ACCESS
Article
Figure 1. Microcrystal morphologies and spectral characterizations
(A) Chemical structures of the synthesized target compound.
(B and C) Corresponding fluorescence microscopy images of two obtained samples.
(D) Absorption (dashed lines), fluorescence (solid lines), and phosphorescence (dotted lines) spectra of monomers (black) in CH2 Cl 2 solutions (1 3
10 5 M), red-emissive (red) and green-emissive (green) microcrystals, respectively.
red emission were obtained when the concentration of the stock CH2Cl2 solution (C)
was 5 mg/mL (Figure 1B), while samples with intense green emission were obtained
at C = 10 mg/mL (Figure 1C). According to scanning electron microscopy (SEM) and
transmission electron microscopy (TEM; Figure S2), both red- and green-emissive
samples are two-dimensional (2D) microribbons (MRs) with smooth surfaces and
sharp edges.
Photophysical properties and structural characterizations
Figure 1D shows the normalized absorption, fluorescence (FL), and phosphores-
cence spectra of red-emissive and green-emissive MRs in comparison with those
of monomers in CH2Cl2 solutions. The related photophysical parameters are sum-
marized in Table 1. The absorption peak of the monomers in dilute solution (1 3
10 5 M) is at 465 nm (Figure 1D, black dashed line). Correspondingly, their FL
peak shows a mirror structure of their absorption, with a maximum emission peak
at 525 nm (Figure 1D, black solid line) at room temperature. The photoluminescence
quantum yield (PLQY) of monomers is determined to be Fmonomers = 1.02% through
an absolute method by using an integration sphere.23 As the variation of solvents’
polarity, the FL emissions of the monomers exhibit strong solvatochromic effects,
indicating CT nature, while their absorption spectra are slightly affected (Figure S3).
In contrast to the monomers, the absorption spectra of the two MRs exhibit an obvi-
ously red-shifted phenomenon, with maximum peaks at 478 nm for green-emissive
MRs and 490 nm for red-emissive MRs. Also, their FL peaks demonstrate significantly
red-shifted emission, with the maxima at 555 and 610 nm for green-emissive and
red-emissive MRs, respectively. Notably, the PLQYs of green-emissive and red-
emissive MRs are also determined to 7.08% and 16.58%, respectively, which are
both much larger than that of the monomers. The substantially red-shifted absorp-
tion and FL peaks and the improved PLQYs of the two MRs are strongly suggestive
of the formation of J-type aggregates with intermolecular CT character in the micro-
crystals. We also carried out phosphorescence spectra of three samples at 77 K (Fig-
ure 1D, dotted lines). Surprisedly, the phosphorescence peaks of three samples
remain almost consistent at 685 nm, indicating that triplet excitons are almost im-
mune to aggregate effects.
Cell Reports Physical Science 3, 100686, January 19, 2022 3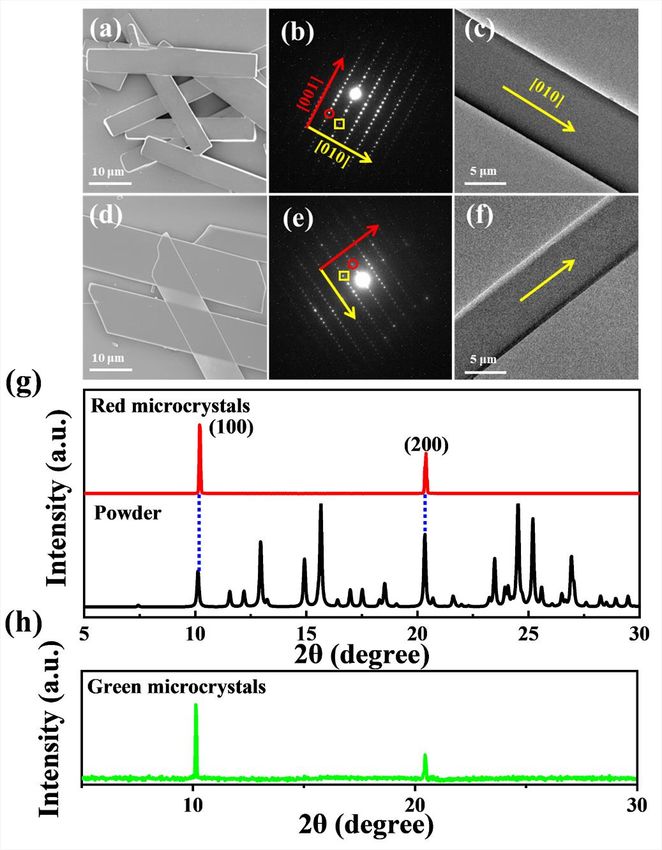
ll
OPEN ACCESS
Article
Table 1. Photophysical parameters of monomers in dilute solution and solid-state microcrystals
1
Sample labs, nm lem, nm tF, ns tDF, ms fF, % fDF, % kr , s
Monomers 465 525 2.65 – 1.02 – 3.85 3 106
Green 478 555 1.58 – 7.08 – 4.48 3 107
Red 490 610 2.23 3.84 16.58 3.61 7.43 3 107
We further measured time-resolved photoluminescence (TRPL) by means of a streak
camera. The emission of the monomers at 525 nm decays monoexponentially,
yielding a lifetime of tm = 2.65 G 0.03 ns at 300 K (Figure S4A). The fluorescence
decay of green-emissive MRs at 555 nm is also fitted monoexponentially, with a
time constant of tgreen = 1.58 G 0.06 ns at 300 K (Figure 2A; Figure S4B), which is
apparently shorter than that of the monomers. No delayed fluorescence was de-
tected for both the monomers and green-emissive MRs. According to the equation
k = PLQY/t, the radiative decay rates (k) are calculated to be km = 3.58 3 106 s 1 and
kgreen = 4.48 3 107 s 1, respectively (Table 1). The 12.5-fold enhancement of k is also
consistent with the J-aggregation model.17 In contrast, the FL decay of red-emissive
MRs at 610 nm shows typically biexponential characteristics, with a short lifetime of
2.23 G 0.05 ns and a long lifetime of 3.84 G 0.01 ms (Figure 2B; Figure S5) in air at
300 K. According to the short time, kred can be calculated to be 7.43 3 107 s 1, which
is much greater than that of the monomers and suggests that red-emissive MRs are
also attributed to J-type aggregation.
It is widely accepted that molecular packing arrangements strongly influence the
excited-state energy levels and optical properties of organic crystals. In order to
understand the packing arrangement of molecules within these two MRs,
selected-area electron diffraction (SAED) and X-ray diffraction (XRD) measure-
ments were carried out. The sharp spots in SAED images reveal that these two
MRs are single crystals. For red-emissive MRs, they belong to monoclinic phase
(P21/c) with lattice parameters of a = 8.7447(17) Å, b = 8.0815(16) Å, c =
23.797(5) Å, a = g = 90 , and b = 93.57 (CCDC No. 2090874; Table S1). Further
analysis of the SAED pattern of a red-emissive MR (Figure S2B) reveals that the sets
of spots marked with the square and circle are due to (010) and (001) Bragg reflec-
tions with d-spacing values of 8.1 and 23.8 Å, respectively, in good agreement
with the cell parameters of a monoclinic crystal structure. Moreover, the XRD curve
(Figure S2G) shows the relative abundance of the crystal planes (100) with d =
8.75 Å. Combining TEM images (Figure S2C), it can be concluded that red-emis-
sive MRs grow preferentially along the crystal [010] direction. Combining the
above analysis with the help of monoclinic crystal structures, molecular packing ar-
rangements within red-emissive MRs are obtained (Figure S6). The molecules are
stacked along the b-axis (i.e., the [010] direction), thereby forming slipped p-stack-
ing, with the shortest p-p separation of about 3.43 Å. The pitch angle between the
molecular transition dipole and the p-stack direction24 is determined to 40 , which
is much smaller than the critical value of 54.7 and might indicate the formation of
J-type aggregation according to Kasha’s exciton model.17,25 Similarly, the squared
and circled sets of spots in the green-emissive MRs correspond respectively to d-
spacing values of 8.5 and 27.4 Å according to SAED pattern (Figure S2E). Further-
more, the XRD is dominated by a series of diffraction peaks with 2q values at
10.136 (and a second peak at 20.448 ) with d = 8.7 Å (Figure S2H). Notably,
the crystalline structural parameters deduced from SAED and XRD measurements
are similar to those of red-emissive MRs, which suggests that green-emissive MRs
might adopt looser molecular packing arrangement compared with red-emissive
MRs.
4 Cell Reports Physical Science 3, 100686, January 19, 2022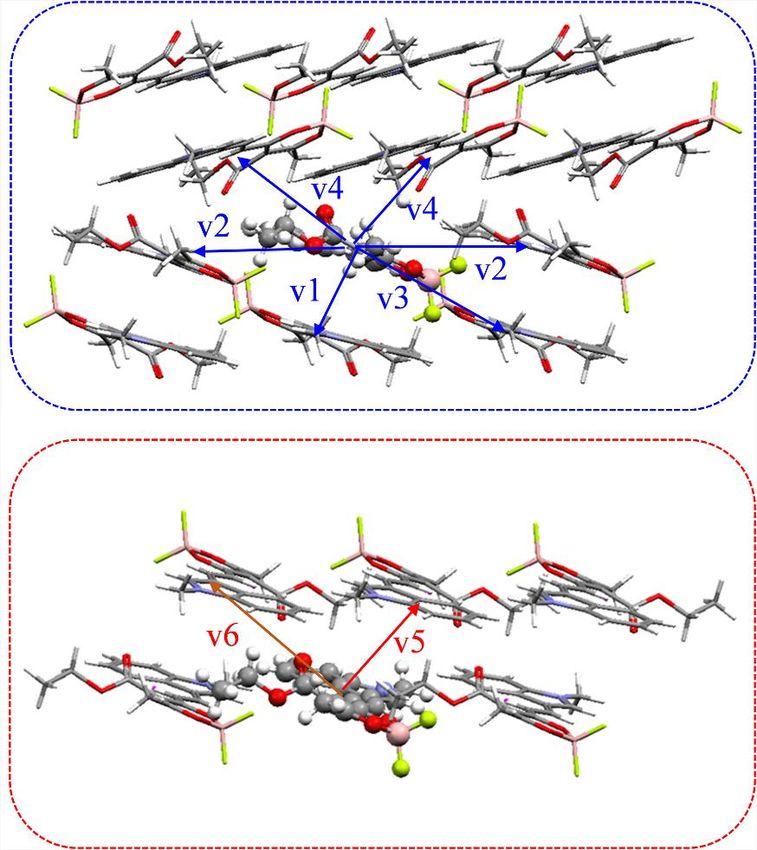
ll
OPEN ACCESS
Article
Figure 2. Time-resolved photoluminescence and temperature-dependent PL spectra and energy
level diagram of two MRs
(A and B) Transient decay curves of green-emissive (A) and red-emissive (B) MRs at 300 K (black line)
and 77 K (red line). Inset in (B): steady-state spectrum (black line) and delay spectrum (red line).
(C and D) PL spectra of green-emissive (C) and red-emissive MRs (D) at the different temperatures.
(E) Energy level diagram for the monomer and two MRs. A, absorption; F, fluorescence; DF, delayed
fluorescence; Phos., phosphorescence.
Theoretical calculations and TADF characterizations
To understand the mechanism of the photophysical behaviors in solid phases, mo-
lecular stacking arrangements in organic crystals are necessary. Because of the
absence of the single-crystal structure of green-emissive MRs, we predict the molec-
ular packing arrangement in green-emissive crystals using the polymorph predictor
module in Materials Studio 6.0 software26–29 and MOMAP 19 software30–34 on the
basis of XRD and SAED results, with reference to the structure of red-emissive
MRs. In order to verify the rationality of predicted molecular stacking, we first calcu-
lated the electronic structures of the monomers and the two MRs (Table S2) by using
density functional theory (DFT) and time-dependent DFT (TDDFT) combined with
the polarizable continuum model (PCM). The calculated energy levels of the mono-
mers are S1 = 2.38 eV and T1 = 1.79 eV, and they are in agreement with the exper-
imental results of the fluorescence spectrum (525 nm) at 300 K and the phosphores-
cence spectrum (685 nm) at 77 K (Figure 1D). In fact, the phosphorescence spectra of
the two MRs were also performed at 77 K (Figure 1D, dotted lines). The phosphores-
cence peaks of both MRs are fully identical to that of the monomers with a similar
decay constant of about 130 ms (Figure S7). Similarly, the calculated energy levels
of the two MRs are determined to S1 = 1.98 eV and T1 = 1.74 eV for red-emissive
Cell Reports Physical Science 3, 100686, January 19, 2022 5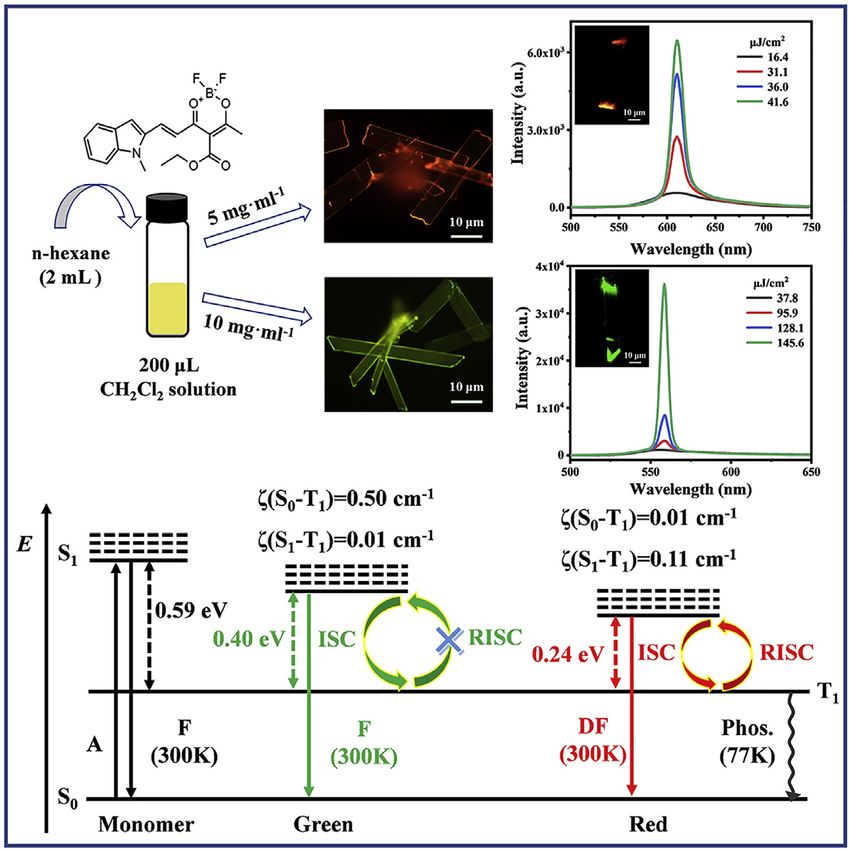
ll
OPEN ACCESS
Article
MRs and S1 = 2.18 eV and T1 = 1.78 eV for green-emissive MRs, which are in agree-
ment with the experimental results (Figure 1D; Figure S7) and suggest the rationality
of molecular arrangements.
We then adopted the energy splitting method for the calculation of the exciton
coupling, which works well for identical dimers,35 on the basis of TDDFT calculation
at the CAM-B3LYP/6-31g(d) level.36 The sign of the coupling is determined by the
aggregation type originating from Kasha’s exciton model,37 that is, negative for J-
type coupling and positive for H-type coupling. The calculated results show that
v1 and v3 dimers among a total of six selected molecular dimers (Figure S8) have
the largest exciton coupling values in red-emissive MRs, while v5 dimer has the
largest exciton coupling value in green-emissive MRs (Table S3). In order to quanti-
tatively measure the intermolecular interactions,38 we calculated the dispersion en-
ergies Edisp using an extended version of the symmetry-adapted perturbation theory
(SAPT) and many-body dispersion (MBD) method (XSAPT+MBD)39 in the Q-Chem
package. It is well known that the smaller the dispersion energy, the stronger the
intermolecular interaction force. From the calculated results (Table S4), v1 dimer
( 25.147 kcal$mol 1) in red-emissive MRs and v5 dimer ( 40.111 kcal$mol 1) in
green-emissive MRs have the strongest intermolecular force and play a dominant
role in the molecular structure. Meanwhile, v1 dimer and v5 dimer both exhibit nega-
tive coupling values, suggesting moderate J-type coupling in both MRs. Combined
with the results of exciton coupling as well as their spectral and structural character-
istics (such as red-shifted spectrum, the spacing of adjacent molecules), it can be
concluded that red-emissive MRs exhibit a higher degree of J-aggregation.
Importantly, red-emissive MRs display significant prompt and delayed photolumi-
nescence (PL) component (Figure 2B), which suggests that efficient TADF properties
are highly expected. To verify the PL origin of the two MRs, we performed temper-
ature-dependent PL and TRPL spectra. As shown in Figure 2C, the PL intensities of
green-emissive MRs gradually increase as the temperature decreases, with an over-
all PL intensity increase of nearly 6 times at 77 K compared with 300 K. This is a typical
fluorescence characteristic. In sharp contrast, the PL spectrum of red-emissive MRs
shows nearly no shift in PL spectra and a decrease in fluorescence intensity by 6
times, along with a decrease in temperature (Figure 2D). This suggests that red-
emissive MRs might be TADF materials, while green-emissive MRs are fluorescence
materials. From the TRPL spectra at 77 K, both the monomers and green-emissive
MRs exhibit longer lifetimes, tm = 7.95 G 0.05 ns and tgreen = 4.86 G 0.09 ns (Figures
S4C and S4D), respectively, which are in good agreement with the mechanism of the
fluorescence emitter. Although red-emissive MRs show prompt and delayed PL
components at 300 K, only a short-lifetime component of 610 nm PL peaks was
left at 77 K, possibly attributable to the blockade of the RISC process. The inset in
Figure 2B shows the spectral results of the time-gated PL measurements on nano-
second-microsecond scale. It is clear that the delayed PL spectra (red line) are
very similar to prompt PL spectra (black line), indicating that they both originate
from the same singlet excited state. This also proves that red-emissive MR is a
TADF emitter.
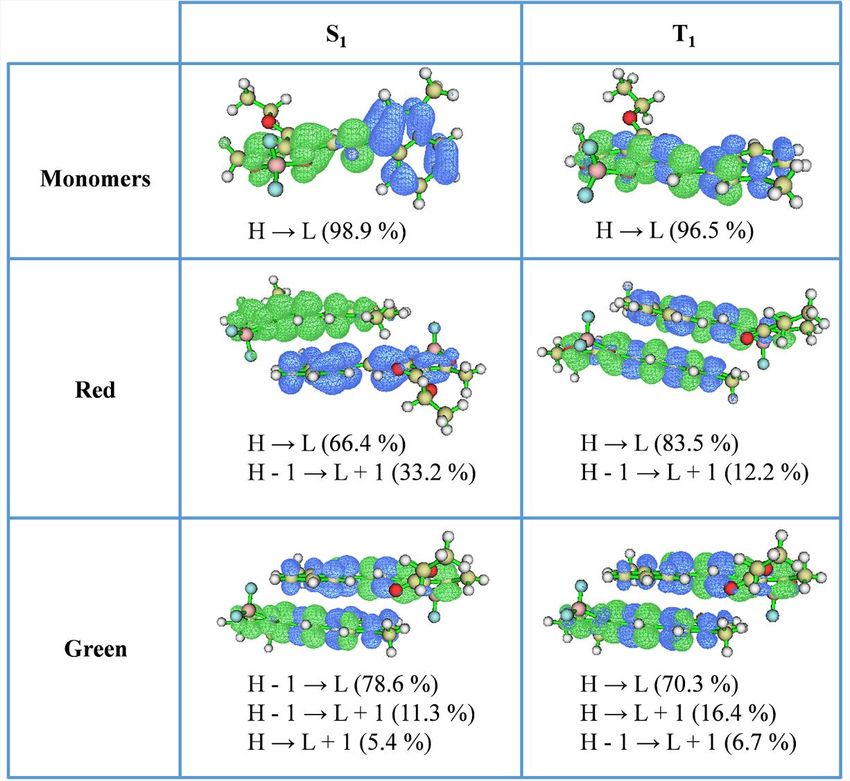
To further understanding the mechanism of TADF activity of the two MRs, the distri-
bution of the HOMOs and LUMOs and analysis of the distribution of the hole and
electron for S1 and T1 states were calculated using the B3LYP/6-31g(d) method
and analyzed by using Multiwfn version 3.5 (Figure S9).40 For the monomers, their
S1 states are predominantly intermolecular CT in nature, while their T1 states demon-
strate a coexistence of local excited state. Meanwhile, the large DEST of 0.59 eV
6 Cell Reports Physical Science 3, 100686, January 19, 2022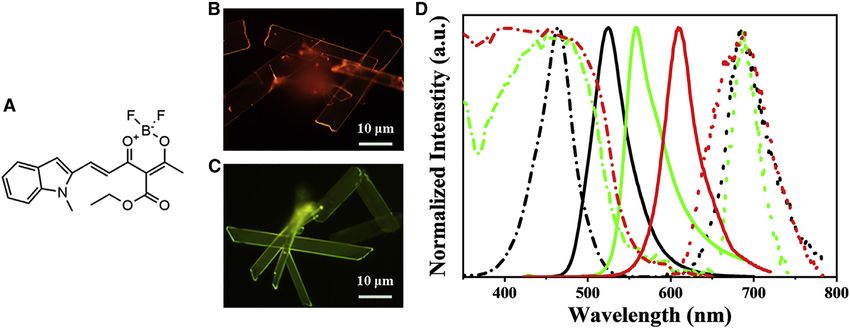
ll
OPEN ACCESS
Article
between S1 and T1 also hinders the RISC process (Figure 2E), such that no TADF ac-
tivity in the monomers is observed. On the basis of the optimized v1 dimer for red-
emissive MRs and v5 dimer for green-emissive MRs, we calculated the transition
configurations of two MRs. Similarly, the S1 states of red-emissive MRs also have a
significant intermolecular CT character, which benefits stabilization of the S1 states.
The smaller DEST of 0.24 eV facilitate to establish the RISC process (Figure 2E).
Although CT nature is unfavorable for molecular dipole transition, J-type aggrega-
tion could enhance electronic dipole transition and contribute large oscillator
strength for efficient PL emission and ASE process.41 In contrast, the S1 and T1 states
of green-emissive MRs both demonstrate a coexistence of LE and CT states. At the
same time, the large DEST of 0.40 eV also impedes the RISC process (Figure 2E), such
that no TADF nature in green-emissive MRs is observed. As is well known, dimers in
crystalline states demonstrate lower S1 energies attributable to the surrounding in-
teractions. For green-emissive MRs, although the S1 energy level is reduced to 2.18
eV, the large DEST of 0.40 eV still obstructs the RISC process (Figure 2E). Meanwhile,
the small spin-orbital coupling (SOC) value of 0.01 cm 1 between S1 and T1 also sug-
gests that the RISC process does not occur in green-emissive MRs. In sharp contrast,
the S1 energy level significantly decreased in red-emissive MRs because of the
tighter molecular packing and higher degree of J-coupling. Only an DEST of 0.24
eV permits an efficient RISC process between S1 and T1. In addition, the SOC value
of 0.11 cm 1 between S1 and T1 is an order of magnitude larger than that of green-
emissive MRs, which also supports an effective RISC process in red-emissive MRs at
300 K. At 77 K, radiative transition from T1 to S0 states generates phosphorescence
because of the absence of the RISC process in red-emissive MRs (Figure 2E).
ASE characterizations
Notably, two MRs both exhibit active waveguiding behaviors (Figures 1B and 1C),
and they may simultaneously serve as active media of PL emission and optical mi-
cro-resonators. Optically pumped lasing measurements were performed on a
homemade micro-PL system (Figure S10). The micro-PL spectra of an isolated sin-
gle MR under different excitation densities (P) of a 400 nm femtosecond laser were
adjusted using a series of neutral density filters. Taking red-emissive MRs as an
example (Figure 3A), a broad spontaneous emission peak at 610 nm is observed
under low pumping density of P < 16.4 mJ/cm2, which exhibits a full width at
half maximum (FWHM) of 56 nm. As P increases from 31.1 to 41.6 mJ/cm2, a strong
ASE emission with FWHM of 11 nm develops around 610 nm and finally becomes
dominant. The emission intensities as a function of P present the expected S-
shaped curve (Figure 3B, blue line): the clear ASE threshold occurs at Pth =
23.6 mJ/cm2, and lasing oscillation starts at P = 36.7 mJ/cm2. Meanwhile, the
FWHM of the PL emission sharply collapses from 56 to 11 nm (Figure 3B, red
line). Together these results demonstrate the transition from spontaneous emis-
sion to ASE processes.42 Similarly, ASE characteristics at 557 nm are also observed
in green-emissive MRs: a sharp collapse of the PL spectra from 33 to 6 nm and a
distinct ASE threshold of 93.2 mJ/cm2 (Figures 3C and 3D).
To further study ASE behaviors of two MRs, TRPL measurements were carried out
for the exploration of the excited-state process of ASE. For green-emissive MRs,
below the ASE threshold (0.6 Pth), a rapid rise followed by nanosecond-scale
monoexponential slow decay with a lifetime constant of about 1.58 G 0.06 ns is
observed (Figure 4A, blue line). In contrast, fast decay with a time constant of
64 G 1.1 ps dominates the decay curve (Figure 4A, red line), which corresponds
to the process of ASE above the threshold (1.5 Pth). We then performed the ASE
threshold as a function of temperature; the experimental results are shown in
Cell Reports Physical Science 3, 100686, January 19, 2022 7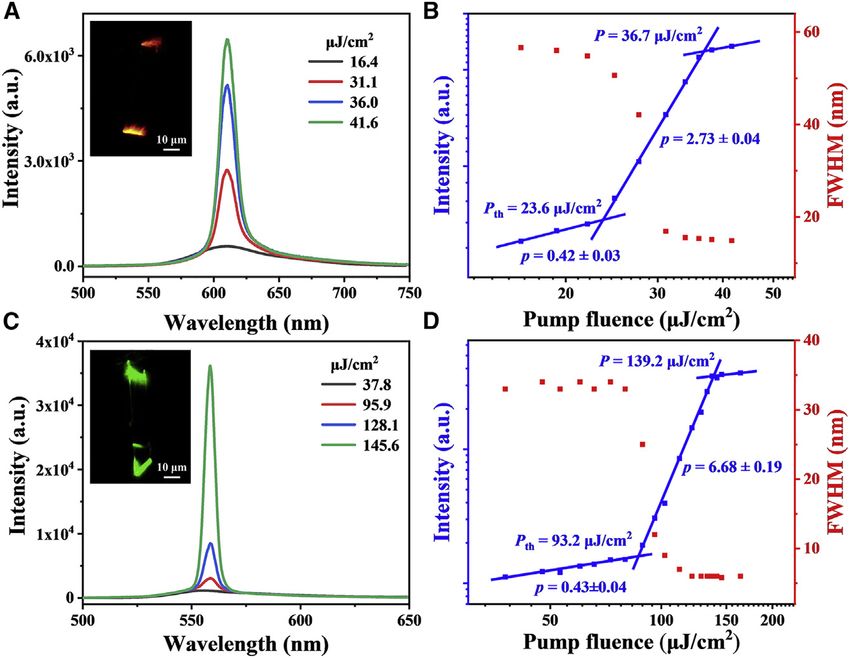
ll
OPEN ACCESS
Article
Figure 3. ASE properties of two MRs
(A and C) PL spectra as a function of pump energy densities in red-emissive (A) and green-emissive
(C) MRs. Inset: corresponding PL images of two microcrystals pumped above the lasing thresholds.
(B and D) Dependence of the peak intensity (blue line) and FWHM of emission spectra (red line) of
red-emissive (B) and green-emissive (D) MRs under different pump fluence.
Figure 4C. As temperature decreases from 300 to 77 K, the ASE threshold of the
single green-emissive MR decreases rapidly by nearly 5 times from about 93.2 to
18.1 mJ/cm2 (Figure 4C, blue line). This phenomenon of the decrease in ASE
threshold as temperature decreases is a typical fluorescence ASE mechanism
based on the fact of forbidden non-radiative transition at low temperature.42
In sharp contrast, the ASE threshold of the single red-emissive MR increase rapidly
exceeded 8 times, from about 23.6 to 210.21 mJ/cm2 (Figure 4C, red line). This result
is completely contradictory to the common fluorescent lasing as observed in green-
emissive MRs. The ASE threshold recovered gradually to its initial value as the tem-
perature was increased to 300 K. This is typical of TADF lasers.15 To further confirm
the ASE from the TADF mechanism in red-emissive MRs, we compare their TRPL
spectra. At low pump fluence (0.4 Pth), the rapid rise followed by monoexponentially
slow decay with a lifetime constant of about 2.23 G 0.05 ns (Figure 4B, blue line),
corresponding to the lifetime of the prompt fluorescence (Figure S5). When the
pump fluence reaches 1.6 Pth, fast decay with a time constant of 99 G 0.8 ps and
a plateau (180 ps) between rapid rise and fast decay were clearly observed (Fig-
ure 4B, red line). The former corresponds to the process of ASE above the ASE
threshold, and the latter indicates that singlet excitons can be supplemented during
the ASE process. In TADF emitters, triplet excitons generated from singlet excitons
through intersystem crossing process can be transformed into singlet excitons by
the RISC process. The regenerated singlet excitons facilitate the establishing of pop-
ulation inversion and slow down the consumption rate of singlet excitons in the ASE
process. When the temperature is decreased to 100 K, the time plateau in the dy-
namic curve totally disappeared because of the inactivation of RISC process (Fig-
ure 4B, green line), which also results in a significant increase in the ASE threshold
8 Cell Reports Physical Science 3, 100686, January 19, 2022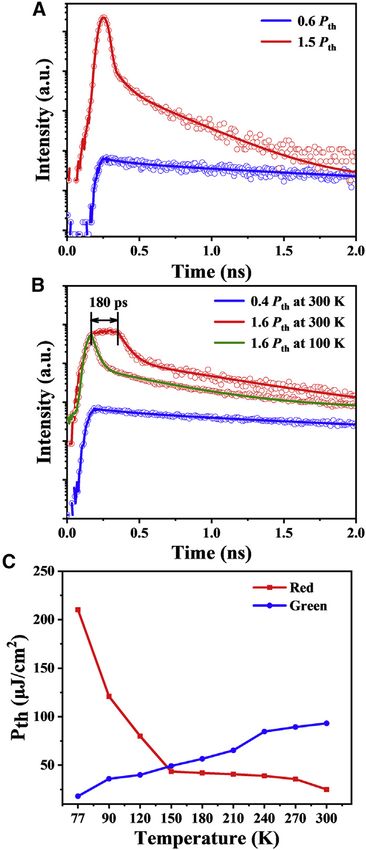
ll
OPEN ACCESS
Article
Figure 4. Comparison of ASE behaviors between fluorescence and TADF
(A) Time-resolved PL decay curves of green-emissive MRs under pump densities of 0.6 Pth (blue line)
and 1.5 Pth at 300 K (red line) monitored at the lasing wavelength using a streak camera.
(B) Time-resolved PL decay curves of red-emissive MRs under pump densities of 0.4 Pth (blue line) at
300 K and 1.6 Pth at 300 K (red line) and 1.6 P th at 100 K (green line) monitored at the lasing
wavelength using a streak camera.
(C) Plot of lasing threshold versa temperature for green-emissive (blue line) and red-emissive (red
line) MRs.
(Figure 4C). Therefore, an effective triplet-mediated TADF ASE in red-emissive MRs
has been demonstrated. Importantly, the larger radiative rate and triplet-assisted
ASE in red-emissive MRs means that their ASE threshold is nearly 4 times lower
than that of green-emissive MRs.
In conclusion, we have demonstrated the regulation of fluorescence and TADF ASE
in organic crystalline polymorphs by controlling the excited-state dynamics. We
design and synthesize a novel organic gain molecule on the basis of the boron di-
fluoride framework and obtain green-emissive and red-emissive crystalline poly-
morphs using a facile solution-assembly method. Experimental and theoretical
studies show that green crystals emit green fluorescence ASE, while red crystals
emit red TADF ASE. Single-crystal data and theoretical simulations indicate that a
Cell Reports Physical Science 3, 100686, January 19, 2022 9ll
OPEN ACCESS
Article
higher degree of J-aggregation in red crystals significantly results in a substantial
decrease in DEST to 0.24 eV for the realization of the RISC process. These results
deepen understanding of the relationship between the aggregation effect and
excited-state dynamics and provide a new strategy for tuning fluorescence ASE
and TADF ASE in pure organic crystals.
EXPERIMENTAL PROCEDURES
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be ful-
filled by the lead contact, Hongbing Fu (hbfu@cnu.edu.cn).
Materials availability
Full details of all materials are provided in the Supplemental information.
Data and code availability
All data associated with this study are included in the article and the Supplemental
information.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.xcrp.
2021.100686.
ACKNOWLEDGMENTS
This work was supported by the National Key R&D Program of China (grants
2018YFA0704805, 2018YFA0704802, and 2017YFA0204503), the National Natural
Science Foundation of China (22150005, 22090022, 21833005, 21873065, and
21790364), the Natural Science Foundation of Beijing (KZ202110028043), the Bei-
jing Talents Project (2019A23), the Open Fund of the State Key Laboratory of Inte-
grated Optoelectronics (IOSKL2019KF01), and Capacity Building for Sci-Tech Inno-
vation-Fundamental Scientific Research Funds, Beijing Advanced Innovation Center
for Imaging Theory and Technology.
AUTHOR CONTRIBUTIONS
Q.L., H.F., and J.Z. initiated and designed the experiments. J.Z. performed all mea-
surements. H.H., L.F., and M.L. performed the calculations. Q.L. and H.F. supervised
the research project and experiments. J.Z., Q.L., C.G., and H.F. wrote the manu-
script. All authors discussed the results and contributed to writing the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Received: August 31, 2021
Revised: October 25, 2021
Accepted: November 17, 2021
Published: December 8, 2021
REFERENCES
1. Eaton, S.W., Fu, A., Wong, A.B., Ning, C.-Z., 2. Röder, R., and Ronning, C. (2018). Review on 3. Chénais, S., and Forget, S. (2012). Recent
and Yang, P. (2016). Semiconductor nanowire the dynamics of semiconductor nanowire advances in solid-state organic lasers. Polym.
lasers. Nat. Rev. Mater. 1, 16028. lasers. Semicond. Sci. Technol. 33, 033001. Int. 61, 390–406.
10 Cell Reports Physical Science 3, 100686, January 19, 2022ll
OPEN ACCESS
Article
4. Ford, E.B., Lystad, V., and Rasio, F.A. (2005). 17. Ye, H., Kim, D.H., Chen, X., Sandanayaka, 30. Shuai, Z., and Peng, Q. (2017). Organic light-
Planet-planet scattering in the upsilon A.S.D., Kim, J.U., Zaborova, E., Canard, G., emitting diodes: theoretical understanding of
Andromedae system. Nature 434, 873–876. Tsuchiya, Y., Choi, E.Y., Wu, J.W., et al. (2018). highly efficient materials and development of
Near-infrared electroluminescence and low computational methodology. Natl. Sci. Rev. 4,
5. Blanche, P.A., Bablumian, A., Voorakaranam, threshold amplified spontaneous emission 224–239.
R., Christenson, C., Lin, W., Gu, T., Flores, D., above 800 nm from a thermally activated
Wang, P., Hsieh, W.Y., Kathaperumal, M., delayed fluorescent emitter. Chem. Mater. 30, 31. Niu, Y., et al. (2018). Molecular Materials
et al. (2010). Holographic three-dimensional 6702–6710. Property Prediction Package (MOMAP) 1.0: a
telepresence using large-area software package for predicting the
photorefractive polymer. Nature 468, 80–83. 18. Reese, C., and Bao, Z. (2007). Organic single- luminescent properties and mobility of organic
crystal field-effect transistors. Mater. Today 10, functional materials. Mol. Physiol. 116, 1078–
6. Kuehne, A.J., and Gather, M.C. (2016). Organic 20–27. 1090.
lasers: recent developments on materials,
device geometries, and fabrication techniques. 19. Kasha, M., Rawls, H.R., and El-Bayoumi, M.A. 32. Shuai, Z., Geng, H., Xu, W., Liao, Y., and André,
Chem. Rev. 116, 12823–12864. (1965). The exciton model in molecular J.M. (2014). From charge transport parameters
spectroscopy. Pure Appl. Chem. 11, 371–392. to charge mobility in organic semiconductors
7. Baldo, M.A., Holmes, R.J., and Forrest, S.R. through multiscale simulation. Chem. Soc. Rev.
(2002). Prospects for electrically pumped 20. Liao, Q., Wang, X.G., Lv, S., Xu, Z., Zhang, Y., 43, 2662–2679.
organic lasers. Phys. Rev. B Condens. Matter and Fu, H. (2018). Cluster-mediated nucleation
Mater. Phys. 66, 035321. and growth of J- and H-type polymorphs of 33. Hsu, C.-P. (2009). The electronic couplings in
difluoroboron avobenzone for organic electron transfer and excitation energy
8. Lehnhardt, M., Riedl, T., Weimann, T., and
microribbon lasers. ACS Nano 12, 5359–5367. transfer. Acc. Chem. Res. 42, 509–518.
Kowalsky, W. (2010). Impact of triplet
absorption and triplet-singlet annihilation on 21. Liao, Q., Wang, Z., Gao, Q., Zhang, Z., Ren, J., 34. Krueger, B.P., Scholes, G.D., and Fleming, G.R.
the dynamics of optically pumped organic De, J., Zhang, X., Xua, Z., and Fu, H. (2018). The (1998). Calculation of couplings and energy-
solid-state lasers. Phys. Rev. B Condens. Matter effect of 1D- and 2D-polymorphs on organic transfer pathways between the pigments of
Mater. Phys. 81, 165206. single-crystal optoelectronic devices: lasers LH2 by the ab initio transition density cube
9. Sandanayaka, A.S.D., Matsushima, T., and field effect transistors. J. Mater. Chem. C method. J. Phys. Chem. B 102, 5378–5386.
Bencheikh, F., Terakawa, S., Potscavage, W.J., Mater. Opt. Electron. Devices 6, 7994–8002.
35. Würthner, F., Kaiser, T.E., and Saha-Möller,
Jr., Qin, C., Fujihara, T., Goushi, K., Ribierre, 22. Jin, X., Huang, H., Wang, X., Liao, Q., Hu, W., C.R. (2011). J-aggregates: from serendipitous
J.C., and Adachi, C. (2019). Indication of and Fu, H. (2020). Control of molecular packing discovery to supramolecular engineering of
current-injection lasing from an organic toward a lateral microresonator for microlaser functional dye materials. Angew. Chem. Int. Ed.
semiconductor. Appl. Phys. Express 12, array. J. Mater. Chem. C Mater. Opt. Electron. Engl. 50, 3376–3410.
061010. Devices 8, 8531–8537.
36. Liu, M., Wei, Y., Ou, Q., Yu, P., Wang, G., Duan,
10. Goushi, K., Yoshida, K., Sato, K., and Adachi, C.
23. Li, S., Fu, L., Xiao, X., Geng, H., Liao, Q., Liao, Y., Y., Geng, H., Peng, Q., Shuai, Z., and Liao, Y.
(2012). Organic light-emitting diodes
and Fu, H. (2021). Regulation of thermally (2021). Molecular design strategy for
employing efficient reverse intersystem
activated delayed fluorescence to room- simultaneously strong luminescence and high
crossing for triplet-to-singlet state conversion.
temperature phosphorescent emission mobility: multichannel CH-p interaction.
Nat. Photonics 6, 253–258.
channels by controlling the excited-states J. Phys. Chem. Lett. 12, 938–946.
11. Di, D., Romanov, A.S., Yang, L., Richter, J.M., dynamics via J- and H-aggregation. Angew.
Rivett, J.P., Jones, S., Thomas, T.H., Abdi Chem. Int. Ed. Engl. 60, 18059–18064. 37. Carter-Fenk, K., Lao, K.U., Liu, K.Y., and
Jalebi, M., Friend, R.H., Linnolahti, M., et al. Herbert, J.M. (2019). Accurate and efficient ab
(2017). High-performance light-emitting 24. Cao, X., Wu, Y., Fu, H., and Yao, J. (2011). Self- initio calculations for supramolecular
diodes based on carbene-metal-amides. assembly of perylenediimide nanobelts and complexes: symmetry-adapted perturbation
Science 356, 159–163. their size-tunable exciton dynamic properties. theory with many-body dispersion. J. Phys.
J. Phys. Chem. Lett. 2, 2163–2167. Chem. Lett. 10, 2706–2714.
12. Uoyama, H., Goushi, K., Shizu, K., Nomura, H.,
and Adachi, C. (2012). Highly efficient organic 25. Spano, F.C. (2010). The spectral signatures of 38. Shao, Y., Gan, Z., Epifanovsky, E., Gilbert,
light-emitting diodes from delayed Frenkel polarons in H- and J-aggregates. Acc. A.T.B., Wormit, M., Kussmann, J., Lange, A.W.,
fluorescence. Nature 492, 234–238. Chem. Res. 43, 429–439. Behn, A., Deng, J., Feng, X., et al. (2014).
Advances in molecular quantum chemistry
13. Hirata, S., Sakai, Y., Masui, K., Tanaka, H., Lee, 26. Rice, B., LeBlanc, L.M., Otero-de-la-Roza, A., contained in the Q-Chem 4 program package.
S.Y., Nomura, H., Nakamura, N., Yasumatsu, Fuchter, M.J., Johnson, E.R., Nelson, J., and Mol. Physiol. 113, 184–215.
M., Nakanotani, H., Zhang, Q., et al. (2015). Jelfs, K.E. (2018). A computational exploration
Highly efficient blue electroluminescence of the crystal energy and charge-carrier 39. Lu, T., and Chen, F. (2012). Multiwfn: a
based on thermally activated delayed mobility landscapes of the chiral [6]helicene multifunctional wavefunction analyzer.
fluorescence. Nat. Mater. 14, 330–336. molecule. Nanoscale 10, 1865–1876. J. Comput. Chem. 33, 580–592.
14. Zhou, Z., Qiao, C., Wang, K., Wang, L., Liang, J., 27. Campbell, J.E., Yang, J., and Day, G.M. (2017). 40. Siebrand, W. (1967). Radiationless transitions in
Peng, Q., Wei, Z., Dong, H., Zhang, C., Shuai, Predicted energy–structure–function maps for polyatomic molecules. II. triplet-ground-state
Z., et al. (2020). Experimentally observed the evaluation of small molecule organic transitions in aromatic hydrocarbons. J. Chem.
reverse intersystem crossing-boosted lasing. semiconductors. J. Mater. Chem. C Mater. Phys. 47, 2411–2422.
Angew. Chem. Int. Ed. Engl. 59, 21677–21682. Opt. Electron. Devices 5, 7574–7584.
41. Li, M., Gao, Q., Liu, P., Liao, Q., Zhang, H., Yao,
15. Li, Y., Wang, K., Liao, Q., Fu, L., Gu, C., Yu, Z., 28. Day, G.M., S Motherwell, W.D., and Jones, W. J., Hu, W., Wu, Y., and Fu, H. (2018). Amplified
and Fu, H. (2021). Tunable triplet-mediated (2007). A strategy for predicting the crystal spontaneous emission based on 2D
multicolor lasing from nondoped organic structures of flexible molecules: the Ruddlesden-Popper perovskites. Adv. Funct.
TADF microcrystals. Nano Lett. 21, 3287–3294. polymorphism of phenobarbital. Phys. Chem. Mater. 28, 1707006.
Chem. Phys. 9, 1693–1704.
16. Nakanotani, H., Furukawa, T., Hosokai, T., 42. Huang, H., Yu, Z., Zhou, D., Li, S., Fu, L., Wu, Y.,
Hatakeyama, T., and Adachi, C. (2017). Light 29. Karfunkel, H.R., and Gdanitz, R.J. (1992). Ab Gu, C., Liao, Q., and Fu, H. (2019). Wavelength-
amplification in molecules exhibiting thermally initio prediction of possible crystal structures turnable organic microring laser arrays from
activated delayed fluorescence. Adv. Opt. for general organic molecules. J. Comput. thermally activated delayed fluorescent
Mater. 5, 1700051. Chem. 13, 1171–1183. emitters. ACS Photonics 6, 3208–3214.
Cell Reports Physical Science 3, 100686, January 19, 2022 11Cell Reports Physical Science, Volume 3 Supplemental information J-aggregation enhanced thermally activated delayed fluorescence for amplified spontaneous emission Jinlong Zhu, Qing Liao, Han Huang, Liyuan Fu, Meihui Liu, Chunling Gu, and Hongbing Fu
Supplemental Figures
1H NMR (600 MHz, Methylene Chloride-d2): δ 8.51 (d, J = 14.9 Hz, 1H), 7.98 – 7.92 (m, 1H),
7.70 (s, 1H), 7.47 (d, J = 1.7 Hz, 1H), 7.46 – 7.44 (m, 1H), 7.43 (dd, J = 7.1, 1.3 Hz, 1H), 7.41 –
7.37 (m, 1H), 4.42 (q, J = 7.2 Hz, 2H), 3.89 (s, 3H), 2.51 (s, 3H), 1.43 (t, J = 7.2 Hz, 3H).
Figure S1. 1H NMR spectrum of compound in deuterated methylene chloride.Figure S2. SEM images of red-emissive (a) and green-emissive MRs (d), respectively. SAED patterns and the corresponding TEM images of single red-emissive (b, c) and green-emissive MRs (e, f), respectively. XRD profiles of red-emissive (g) and green-emissive MRs (h) grown on silicon substrates, respectively.
Figure S3. The steady-state absorption (a) and PL spectra (b) of monomers in the different solvents. Note: Abbreviations for different solvents: toluene (TOL), butyl ether (BE), ethyl ether (EE), ethyl acetate (EA), tetrahydrofuran (THF), dichloromethane (DCM), acetone (ACE), and acetonitrile (ACN).
Figure S4. Transient decay spectra of the monomers (a,c) and green-emissive MRs (b,d).
Figure S5. Transient decay spectra of red-emissive MRs at 300 K (a,b) and 77 K (c).
Figure S6. Molecular packing arrangement of the molecules in red-emissive MRs: view of the long axis of the molecule with the illustration of the pitch angle. (b) View of the short axis of the molecule with the illustration of the roll angle. According to this figure, the pitch angle and the longitudinal displacement are determined to 40° and 4.61 Å while the roll angle and transverse displacement are 52° and 2.58 Å, respectively. If a π-stack undergoes a roll angle of 45°, the adjacent molecule slide is far more than the width of aromatic rings (~3 Å), which will destroy the overlap between the π-orbitals of adjacent molecules. Here, the roll angle of roughly 52° in red-emissive MRs severely influences the π-π interaction of adjacent molecules, resulting in almost no π-π interaction between molecules.1
Figure S7. (a) Phosphorescence emission spectra of the monomers in dilute CH 2Cl2 solution (black), red-emissive (red) and green-emissive MRs (green) at 77 K. Transient decay spectra of monomers in dilute CH2Cl2 solution (b), red-emissive (c) and green-emissive MRs (d).
Figure S8. The accumulation of all dimers in the red-emissive MRs.
Figure S9. The distribution of the HOMOs and LUMOs and the analysis for the distribution of the hole (blue) and electron (green) for S1 and T1.
Figure S10. Schematic demonstration of the experimental setup for the optical characterization: (a) the confocal optical microscopy and (b) the transmittance optical path for the waveguide measurements. The second harmonic (λ = 400 nm, pulse width 150 fs) of a 1 kHz Ti:sapphire regenerative amplifier was focused to a 50 μm diameter spot to uniformly excite the selected isolated MR on a 2D movable table. The pump density (P) of excitaion laser is adjusted by using neutral density filters. Spatially resolved PL spectra were collected underneath by using a three dimensional movable objective (50 × 0.9 NA) coupled to an optical fiber and detected using a liquid-nitrogen-cooled charge-coupled device (CCD).
Supplemental Tables:
Table S1. Crystallographic data for Red single crystal.
Identification code Red
Empirical formula C18H18BF2NO4
Formula weight 361.14
Crystal system monoclinic
Space group P21/c
a/Å 8.7447(17)
b/Å 8.0815(16)
c/Å 23.797(5)
α/° 90
β/° 93.57(3)
γ/° 90
Volume/Å3 1678.5(6)
Z 4
ρcalcg/cm3 1.429Table S2. Calculated and experimental photophysical parameters of monomers in dilute
solution and solid-state MRs.
a c b d
Sample S 1 (eV) S 1 (eV) T (eV) T (eV) ΔEST (eV)
1 1
Monomers 2.36 2.38 1.81 1.79 0.59
Green-emissive MRs 2.23 2.18 1.81 1.78 0.40
Red-emissive MRs 2.03 1.98 1.81 1.74 0.24
Note: Experimental photophysical parameters of emission energy of singlet (a) and triplet (b).
Calculated adiabatic energy levels of singlet (c) and triplet (d). ΔEST : energy gaps between
singlet and triplet.Table S3. Exciton coupling values and dimer packing parameters in red-emissive and
green-emissive MRs.
Dipole moment
dc-c (Å) J (meV) θ (°)
displacement (Å)
v1 6.019 -83.03 40 4.61
v2 8.081 55.44 64 3.54
v3 9.430 87.65 83 1.15
v4 7.110 -1.23 35 5.82
Red
v5 10.443 -54.19 29 9.13
v6 13.713 -29.65 22 12.71
v1 6.181 -72.09 38 4.87
v2 8.500 47.34 64 3.73
v3 9.726 76.13 78 2.02
v4 7.110 -1.23 33 5.96
Green
v5 11.710 -80.19 25 10.61
v6 15.587 -22.44 18 14.82
We used the energy splitting method to calculate the coupling between neighbored
molecules, which works well for identical molecular dimer. This method will include the
coupling from Förster (Coulomb) mechanism and Dexter (exchange) mechanism both, where
the later component will be missed by dipole-dipole interaction method or transition density
cube method. So when the intermolecular distance is small, this method will be more reliable.
For large molecular distance, the coupling was calculated by dipole-dipole interaction. The
calculation is based on the TDDFT calculation at CAM-B3LYP/6-31g(d) level.Table S4. XSAPT+MBD energy decomposition of all dimers of red-emissive and
green-emissive MRs.
Edisp Eelst Eexch Eind Eexch-ind
v1 -25.147 -8.488 17.123 -8.736 5.632
v2 -10.114 -1.418 4.831 -3.122 0.914
Red v3 -9.766 -14.921 7.593 -6.703 1.842
v4 -14.020 -4.064 8.624 -3.566 1.898
v5 -10.145 -4.859 6.173 -2.839 1.632
v6 -3.380 0.075 1.669 -0.472 0.270
v1 -21.412 -6.156 11.887 -5.888 3.549
v2 -6.631 -0.551 1.686 -1.952 0.289
Green v3 -7.902 -14.121 6.990 -5.545 1.706
v4 -14.020 -4.064 8.624 -3.566 1.898
v5 -40.111 -51.675 148.621 -79.828 64.010
v6 -0.613 0.592 0.003 -0.074 0.350
Note: All energetics are shown in the unit of kcal•mol-1.
Edisp : dispersion energies
Eelst : electrostatic forces
Eexch : exchange electrostatic term
Eind : induced force
Eexch-ind : exchange induced term
In order to quantitatively measure the intermolecular interactions, we calculated the
dispersion energies Edisp by using an extended version of the symmetry-adapted perturbation
theory (SAPT) and many-body dispersion (MBD) method (XSAPT+MBD) in Q-Chem package.Supplemental Notes
Note S1. Synthesis procedure of the organic difluoroboron derivative.2
In a 50 mL flask, the mixture of ethyl diacetoacetate (228 μL, 1.463 mmol) and BF3∙Et2O
(199 μL, 1.609 mmol) in 3 mL ethyl acetate was heated for 30 min at 50-60 ℃ in air.
1-methyl-1H-indole-3-carbaldehyde (0.582 g, 3.658 mmol) and B(n-OBu)3 (0.987 mL, 3.658
mmol) were dissolved into 12 mL ethyl acetate, and then the solution was injected into the first
mixture. The reaction was kept at 50-60 ℃ for another 30 min. First portion of BuNH 2 (58 μL,
0.585 mmol) was added dropwise into the reaction. After 6 h heating, second portion of BuNH2
(29 μL, 0.293 mmol) was added, and the reaction was kept heating at 50-60 ℃ overnight. All
the solvents were evaporated. The crude product was obtained by flash column
chromatography (silica, dichloromethane (CH2Cl2)) mixed with few ligand and aldehyde. The
further purification was done by multiple precipitations in CH 2Cl2/petroleum ether mixtures.
Scheme S1. The synthetic route of the compound.Supplemental Experimental Procedures Materials All the chemicals and reagents were purchased from commercial sources and used as received without further purification. The molecule synthesized was purified by column chromatography and recrystallization from dichloromethane and petroleum for two times, and fully characterized by 1H NMR and high resolution mass spectroscopies (HRMS). 1H NMR (400 MHz) spectra were recorded in deuterated solvents on a Bruker ADVANCE 400 NMR Spectrometer. Tetramethylsilane (TMS) was used as the internal standard. HRMS were recorded on a GCT premier CAB048 mass spectrometer operating in MALDI-TOF mode. Steady-state spectroscopic measurements The UV-visible absorption spectra were measured on a Shimadzu UV-3600 spectrometer with a slit width of 1 nm. The fluorescence emission spectroscopy and absolute luminescence quantum yield were measured on a Hitachi F-4600 spectrophotometers equipped with a xenon arc lamp and an integrating sphere. The lifetime and time-resolved emission spectroscopic measurement We built the picosecond time resolution device independently for testing, the device configuration is as follows: the second harmonic (400 nm, 150 fs, 1 kHz) of a regenerative amplifier (Spitfire, Spectra Physics) seeded with a mode-locked Ti:sapphire laser (Tsunami, Spectra Physics) was used to excite the samples at the front surface. In order to avoid interference as much as possible, the excitation pulse light and the signal light are spatially perpendicular to each other. The signal collected along the direction normal to the sample surface was dispersed by a polychromator (250is, Chromex) and detected with a streak camera (C5068, Hamamatsu Photonics). The temporal resolution was slightly different depending on the delay width used. X-Ray Crystallography The single crystal sample of red was achieved from slowly evaporative crystallization using n-hexane/dichloromethane (3:1, v/v). Single crystal data was collected on a SuperNova, Dual, AtlasS2 diffractometer using graphite monochromated CuKα radiation (λ = 1.54178 Å). The crystal was kept at 100.00 (10) K during data collection. Using Olex2, the structure was solved with the ShelXT structure solution program using Intrinsic Phasing and refined with the ShelXL refinement package using Least Squares minimization. Home-made micro-PL system The second harmonic (λ = 400 nm, pulse width 150 fs) of a 1 kHz Ti:sapphire regenerative amplifier was focused to a 50 μm diameter spot to uniformly excite the selected isolated MR on a 2D movable table. The pump density (P) of excitation laser is adjusted by using neutral density filters. Spatially resolved PL spectra were collected underneath by using a three dimensional movable objective (50 × 0.9 NA) coupled to an optical fiber and detected using a liquid-nitrogen-cooled charge-coupled device (CCD).
Supplemental References
1 Liao, Q. et al. (2018). The effect of 1D- and 2D-polymorphs on organic single-crystal
optoelectronic devices: lasers and field effect transistors. J. Mater. Chem. C 6,
7994-8002.
2 Kim, D.-H. et al. (2018). High-efficiency electroluminescence and amplified
spontaneous emission from a thermally activated delayed fluorescent near-infrared
emitter. Nature Photonics 12, 98-104.You can also read

















































