GENETIC CODE EXPANSION IN NEURONS FOR STUDYING PATHOGENIC MECHANISMS OF ALZHEIMER DISEASE
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
From the Division of Neurogeriatrics,
Department of Neurobiology, Care Sciences and Society,
Karolinska Institutet, Stockholm, Sweden
GENETIC CODE EXPANSION IN
NEURONS FOR STUDYING PATHOGENIC
MECHANISMS OF ALZHEIMER DISEASE
Lea Susanne van Husen
Stockholm 2022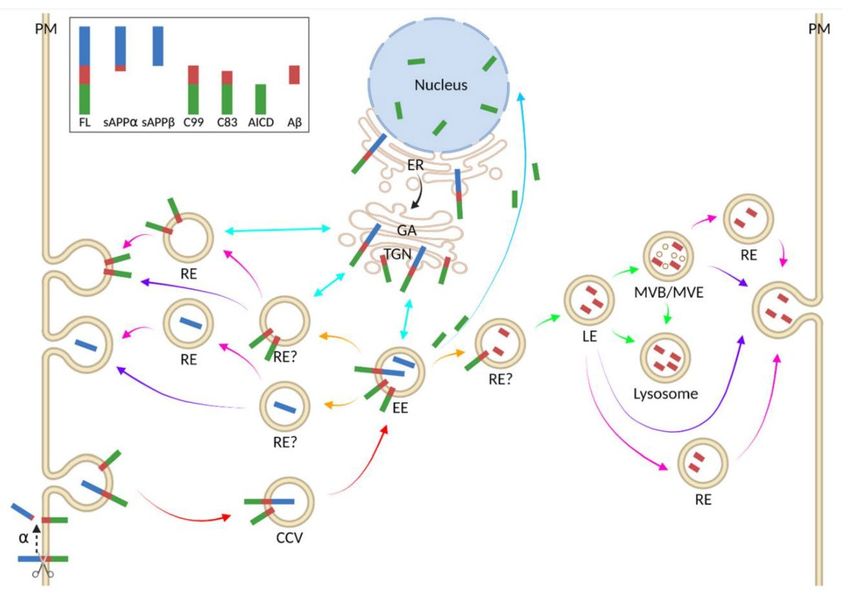
All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by Universitetsservice US-AB, 2022 © Lea Susanne van Husen, 2022 ISBN 978-91-8016-514-3

Genetic code expansion in neurons for studying
pathogenic mechanisms of Alzheimer disease
THESIS FOR DOCTORAL DEGREE (Ph.D.)
By
Lea Susanne van Husen
The thesis will be defended in public at SciLifeLab in Air and Fire, Tomtebodavägen 23 A
Solna, Sweden, Friday the 25th of February at 9.00.
Principal Supervisor: Opponent:
Assoc. Prof. Sophia Schedin Weiss Dr. Ivana Nikić-Spiegel
Karolinska Institutet Eberhard Karls Universität Tübingen
Department of Neurobiology, Care Molecular Mechanisms of Axonal Injury
Sciences and Society Werner Reichardt Centre for Integrative
Division of Neurogeriatrics Neuroscience
Co-supervisor(s): Examination Board:
Assoc. Prof. Lars O. Tjernberg Prof. Nico Dantuma
Karolinska Institutet Karolinska Institutet
Department of Neurobiology, Care Department of Cell and Molecular
Sciences and Society Biology
Division of Neurogeriatrics
Prof. Ola Hermanson
Assoc. Prof. Simon J. Elsässer Karolinska Institutet
Karolinska Institutet Department of Neuroscience
Department of Medical
Biochemistry and Biophysics Docent Anna-Lena Ström
Division of Genome biology Stockholm’s Universitet
Department of Biochemistry and
Prof. Bengt Winblad Biophysics
Karolinska Institutet
Department of Neurobiology, Care
Sciences and Society
Division of Neurogeriatrics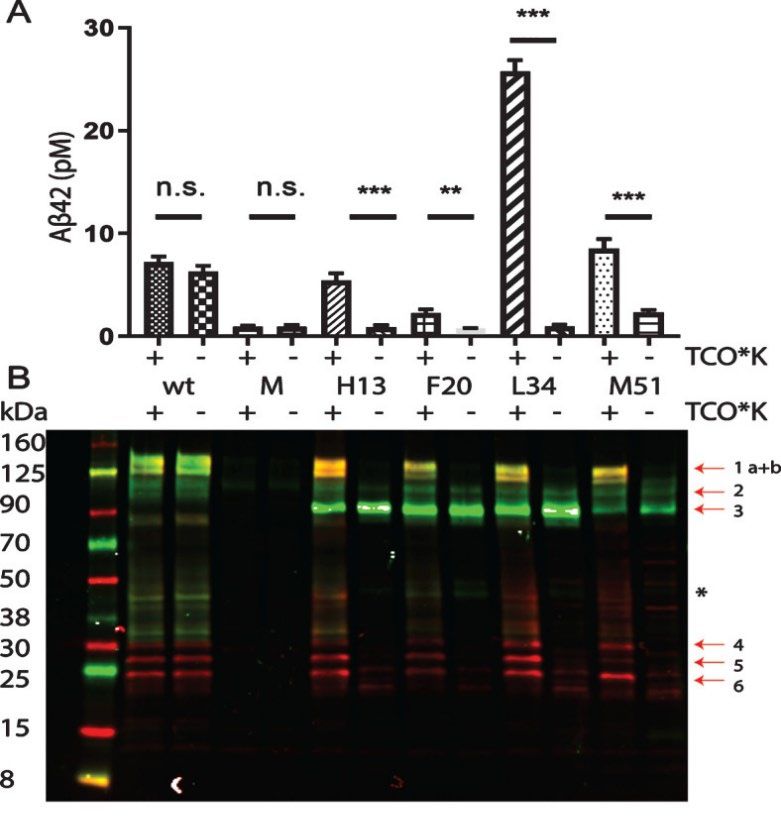
To my family.
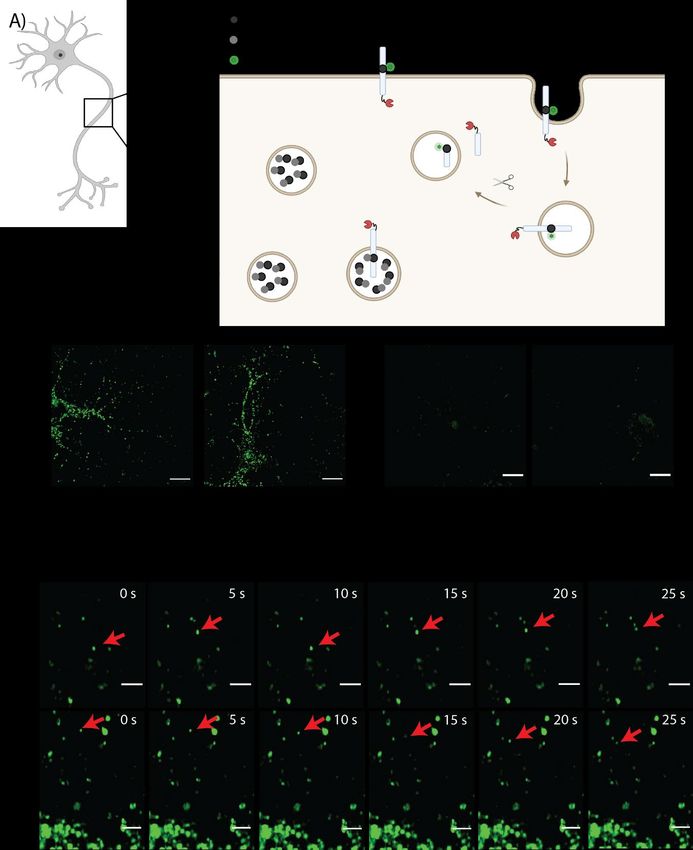
POPULAR SCIENCE SUMMARY OF THE THESIS How to hack the genetic code of neurons? What sounds like one of Dr. Frankenstein’s ideas is actually a way to understand molecular processes in the brain and can help us to study neurodegenerative diseases like Alzheimer disease. Every three seconds a new patient is diagnosed with Alzheimer’s disease. A key player is the Amyloid Precursor Protein, also called APP. It gets processed in the neurons and a resulting fragment accumulates in the brain and causes neurotoxic effects. I want to study that protein, but therefore I need precious tools and a good model system, since it is extremely difficult to observe the human brain in action. One solution can be to study Minibrains – also called cerebral organoids. They can be made from undifferentiated human induced pluripotent stem cells, which we differentiate to neurons. Since they can be made from skin cells nobody needs to donate their neurons, we just need a small skin biopsy. Okay so far, but why do we need to hack the genetic code of the Minibrains? Because we want to add unnatural amino acids! When we add an unnatural amino acid instead of a natural one, we can label proteins and small peptides in living cells and the changes we make in the protein are very small. This enables us to study protein transport and molecular processes in the neurons without influencing it. But how do we do it? Three of the nucleotide triplets that translate mRNA into proteins are used as stop codons to mark where the protein ends instead of encoding for an amino acid. The least used stop codon in mammalian cells is the amber codon. I mutated one codon in the DNA that encodes my protein of interest to this stop codon. But since I don’t want the translation to stop there, I also added a bunch of cellular components. We need tRNA, that recognizes the amber codon and an enzyme that recognizes the unnatural amino acids and pair them with the tRNAs. With these components the translation does not stop at the stop codon, but the unnatural amino acid gets incorporated, and the translation continues till the whole protein is produced.
In the figure above you can see a living organoid expressing the green fluorescent protein GFP with an amber codon in the middle. I also enabled the incorporation of the unnatural amino acids in APP in a cell line. In the future, it would be great to follow APP and its processing products in organoids to detect cellular processes that we can’t study in animals - since a mouse brain is not a human brain. I hope you are curious to learn more about genetic code expansion in this thesis and are not afraid anymore that I created a new Frankenstein’s monster.
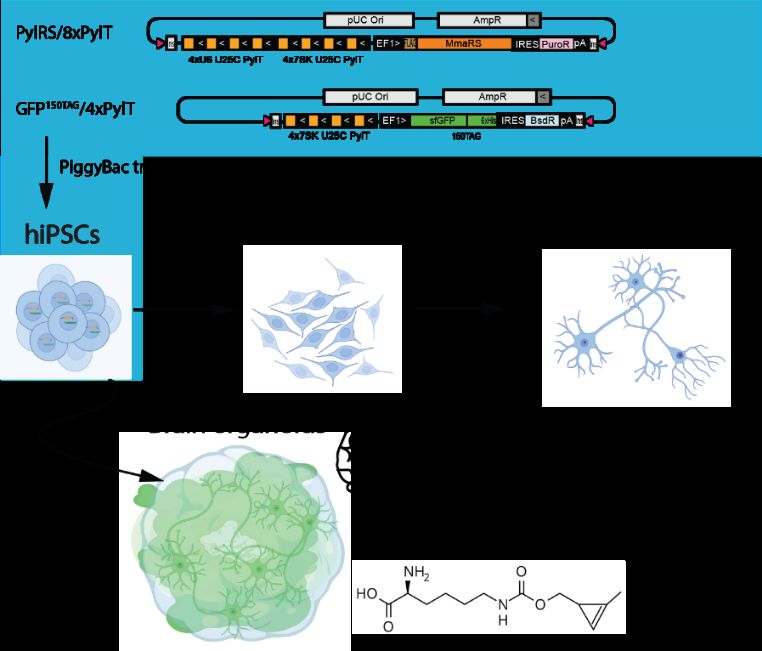
ABSTRACT Every three seconds a new patient is diagnosed with Alzheimer disease (AD). The primary risk factor for this irreversible, progressive brain disorder is old age. It is a growing health concern, not only for the patients, but also for their relatives and society, because the number of affected people is expected to double every 20 years. New tools are necessary to expand the knowledge about the molecular mechanisms behind AD. The small neurotoxic amyloid β-peptide (Aβ) is produced by processing of the Amyloid Precursor Protein (APP) through a cascade of secretase cleavages. However, it is not fully understood in which subcellular location the processing takes place and how this is changed during the course of the disease. A major problem is the lack of methods labeling small peptides like Aβ in living cells. To investigate where the processing takes place, we developed a dual fluorescence labeling system. The C-terminal tail of APP is fluorescently labeled using a SNAP-tag, while the Aβ region of APP is fluorescently tagged with a turn-on dye at a non-canonical amino acid (ncAA). The ncAA is introduced at specific positions in APP using a genetic code expansion (GCE) strategy. Using this approach, APP can be fluorescently labeled at two sites in living HEK293T cells with minimal background. However, a key to understand the underlying molecular mechanisms in AD are appropriate model systems. Therefore, we produced Baculoviruses to transduce primary cortical neurons from mice with the GCE machinery and APP with a SNAP- tag and an amber codon. We reduced background labeling caused by tetrazine dyes in the neurons by developing a two-step labeling protocol to block excessive ncAA. In parallel, we also established GCE in human induced pluripotent stem cells (hiPSCs) and differentiated them to neurons and brain organoids. The stable hiPSCs expressing the GCE machinery provide a unique platform to study and manipulate proteins in different cell types and development stages.
LIST OF SCIENTIFIC PAPERS
I. Lea S. van Husen, Sophia Schedin-Weiss, Minh Nguyen Trung, Manija A.
Kazmi, Bengt Winblad, Thomas P. Sakmar, Simon J. Elsässer, and Lars O.
Tjernberg
Dual Bioorthogonal Labeling of the Amyloid-β Protein Precursor Facilitates
Simultaneous Visualization of the Protein and Its Cleavage Products. J
Alzheimers Dis. 2019;72(2):537–548
II. Lea S. van Husen, Simon J. Elsässer, Lars O. Tjernberg and Sophia Schedin-
Weiss
Genetic Code Expansion for fluorescence labeling of APP in primary neurons
manuscript
III. Lea S. van Husen, Anna-Maria Katsori, Birthe Meineke, Lars O. Tjernberg,
Sophia Schedin-Weiss, Simon J. Elsässer
Engineered human induced pluripotent cells enable genetic code expansion
in brain organoids
ChemBioChem 2021,22, 3208–321CONTENTS
1 INTRODUCTION...........................................................................................................1
2 LITERATURE REVIEW ...............................................................................................3
2.1 Genetic Code Expansion .......................................................................................3
2.1.1 Protein biosynthesis ..................................................................................3
2.1.2 Natural expansion of the genetic code ......................................................4
2.1.3 Amber codon suppression .........................................................................5
2.1.4 Advantages of amber codon suppression .................................................8
2.1.5 Limitations of amber codon suppression..................................................9
2.2 Bioorthogonal labeling ........................................................................................13
2.2.1 SNAP-tag labeling ..................................................................................13
2.2.2 SPIEDAC reaction ..................................................................................14
2.3 Genetic code expansion for neuronal studies .....................................................17
2.3.1 Genetic code expansion in cell lines.......................................................17
2.3.2 Genetic code expansion in primary neurons ..........................................18
2.3.3 Genetic code expansion in vivo ..............................................................19
2.3.4 Genetic code expansion in human cells..................................................20
2.3.5 Genetic code expansion to study AD .....................................................20
2.4 Alzheimer’s Disease ............................................................................................21
2.4.1 An introduction to Alzheimer’s disease .................................................21
2.4.2 APP Processing and Transport ...............................................................22
2.4.3 Pathological and physiological effects of APP fragments .....................25
2.4.4 Treatments targeting APP Processing or Aβ ..........................................26
2.5 Models for APP processing .................................................................................27
2.5.1 Human Embryonic Kidney 293 (HEK293) cells ...................................27
2.5.2 Primary neurons from mice ....................................................................27
2.5.3 Human induced pluripotent stem cells ...................................................28
3 RESEARCH AIMS .......................................................................................................31
4 MATERIALS AND METHODS .................................................................................33
4.1 Non-canonical amino acids .................................................................................33
4.2 Constructs, cloning and mutagenesis ..................................................................33
4.3 Cell culture...........................................................................................................34
4.3.1 HEK293T cells ........................................................................................34
4.3.2 Primary cortical neurons .........................................................................34
4.3.3 Human induced pluripotent stem cells ...................................................34
4.3.4 Neural stem cells .....................................................................................35
4.3.5 Neurons derived from hiPSCs ................................................................35
4.3.6 Differentiation of human induced pluripotent stem cells to
neuronal stem cells and neurons .............................................................35
4.3.7 Generation of stable organoids ...............................................................35
4.4 Transfection .........................................................................................................35
4.4.1 Transient transfection of HEK293T cells ...............................................364.4.2 Transient transfection of primary cortical neurons ................................ 36
4.4.3 Generation of stable human induced pluripotent stem cells .................. 36
4.5 Generation of Baculoviruses and transduction ................................................... 36
4.5.1 Generation of Baculoviruses................................................................... 36
4.5.2 Transduction of HEK293T cells and neurons ........................................ 37
4.6 Bioorthogonal labeling ........................................................................................ 37
4.6.1 Labeling of HEK293T cells .................................................................... 37
4.6.2 Labeling of neurons derived from hiPSCs ............................................. 37
4.6.3 Labeling of primary neurons................................................................... 37
4.7 Analytical techniques for detection of proteins .................................................. 38
4.7.1 Immunofluorescence ............................................................................... 38
4.7.2 Light-sheet microscopy and immunofluorescence microscopy ............ 39
4.7.3 Western blotting ...................................................................................... 39
4.7.4 Determination of Aβ42 levels by ELISA ............................................... 40
4.7.5 Flow Cytometry ...................................................................................... 40
5 RESULTS AND DISCUSSION................................................................................... 41
5.1 PAPER I: Dual Bioorthogonal Labeling of the Amyloid-β Protein
Precursor Facilitates Simultaneous Visualization of the Protein and Its
Cleavage Products ............................................................................................... 41
5.2 PAPER II: Genetic Code Expansion for fluorescence labeling of APP in
primary neurons ................................................................................................... 44
5.3 PAPER III: Engineered Human Induced Pluripotent Cells Enable Genetic
Code Expansion in Brain Organoids .................................................................. 47
6 CONCLUSIONS ........................................................................................................... 49
7 FUTURE PERSPECTIVES.......................................................................................... 51
8 ACKNOWLEDGEMENTS.......................................................................................... 53
9 REFERENCES .............................................................................................................. 57LIST OF ABBREVIATIONS A Adenine AD Alzheimer Disease ADAM A Disintegrin and Metalloprotease AICD APP intracellular domain AMPA α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid APH-1 Anterior pharynx defective 1 APOE Apolipoprotein E APP Amyloid precursor protein ATTO488 Tetrazine-ATTO-488 Aβ Amyloid β-peptide BDPFL 6-methyl-tetrazine-BODIPY-FL BCNK Bicyclo[6.1.0] nonyne - Lysine BONCAT Bioorthogonal ncAA tagging BzF 4-Benzoyl - L - phenylalanine C Cytosine CCV Clathrin-coated vesicle. CpK Cyclopropene - L - Lysine CTF Carboxyterminal fragments CuAAC Copper catalyzed azide-alkyne 1,3-dipolar cycloaddition EE Early endosome EF-Tu Elongation factor thermo unstable ER Endoplasmic reticulum eRF1 Eukaryotic release factor 1 FAD Familial AD FRET Förster resonance energy transfer G Guanine GA Golgi apparatus GCE Genetic code expansion hAGT O6-alkylguanine-DNA alkyltransferase hiPSCs Human induced pluripotent stem cells
LE Late endosome
mRNA messenger RNA
MVB Multivesicular body
MVE Multivesicular endosome
NcAA Non-canonical amino acid
Nct Nicastrin
NMDA N-methyl-D-aspartate
PEN2 Presenilin enhancer 2
PM Plasmamembrane
PS Presenilin
Pyl Pyrrolysine
PylRS Pyrrolysyl-tRNA synthetase
RE Recycling endosome
SAD Sporadic AD
sAPP sAPP
Sec Selenocysteine
SECIS Sec insertion sequence
SPAAC Strain promoted azide-alkyne cycloaddition
SPIEDAC Strain-promoted inverse-electron-demand Diels–Alder
cycloaddition
Thymine
T
Cyclooct - 2 - en - L - Lysine
TCOK
Trans-Golgi network
TGN
Transfer-RNA
tRNA
Uracil
U
Vesicular stomatitis virus G-glycoprotein
VSVG1 INTRODUCTION
New tools in our toolbox for studying biochemical processes in neuroscience are essential to
understand fundamental mechanisms in our brain and to shed light on changes that occur in
our neurons due to diseases or age. Since my thesis is mainly focusing on what I did during my
PhD studies, I want to use this first page to answer the question: Why? Why did I want to work
on expanding the genetic code to study Alzheimer’s disease (AD)?
Every three seconds a new patient is diagnosed with AD. The primary risk factor for the
irreversible, progressive brain disorder is old age. It is a growing health concern, not only for
the patients, but also for their relatives and society. However, the pathological mechanisms
remain to be determined. Understanding the underlying mechanisms, like how the amyloid
precursor protein (APP) is transported and processed in neurons, is essential to understand the
changes that occur and to develop drugs to mitigate the symptoms or even cure the disease.
A major problem in studying AD is the lack of methods labeling small peptides like the
neurotoxic amyloid β-peptide (Aβ) in living cells. Therefore, we want to address the problem
by establishing a genetic code expansion (GCE) approach in primary neurons from mice and
neurons derived from human induced pluripotent stem cells (hiPSCs). This method enables us
to label small peptides specifically at selected positions in the protein, without altering the
properties of the peptide.
However, this method can also be used to study other diseases and processes than AD. Through
paper III, where we engineered hiPSCs to enable GCE in brain organoids, we hope to provide
a protocol to study a wide range of molecular mechanisms in living human cells. HiPSCs can
be differentiated to different cell types. Therefore, I hope that this work provides a small step
towards adding a new tool in our toolbox to manipulate proteins and studying molecular
mechanisms.
12
2 LITERATURE REVIEW
2.1 GENETIC CODE EXPANSION
2.1.1 Protein biosynthesis
A code is a system to translate information from one representation into another form of
representation. In case of the genetic code, the information is encoded in the double-stranded
DNA molecule made from nucleotide building blocks with four different nucleobases: adenine
(A), thymine (T), guanine (G) and cytosine (C). In the nucleus, the DNA sequence is
transcribed into messenger RNA (mRNA), which also consists of nucleotides, although with
thymine replaced by uracil (U). The mRNA is exported from the nucleus and used as a template
for protein biosynthesis in the cytoplasm. Proteins are polymers composed of their building
blocks, which are called amino acids. Francis Crick, Sydney Brenner, Leslie Barnett and R.J.
Watts-Tobin demonstrated that three nucleotides are always needed to encode for one amino
acid 1. Through subsequent work by Robert W. Holley, Har Gobind Khorana, Marshall W.
Nirenberg and others, we now know that 61 of the possible 64 triplet codons (43 = 64) decode
20 canonical amino acids (these are also termed ‘sense’ codons), while three codons (amber
(UAG), opal (UGA) and ochre (UAA)) are stop codons (‘nonsense’ codons) 2. Sense codons
are decoded by transfer RNAs (tRNAs) which carry a complementary or near-complementary
nucleobase triplet in their anticodon loop and the corresponding amino acid esterified to their
3’ end. Between one and six different codons specify a single amino acid via one or several
distinct tRNAs. Therefore, the genetic code is redundant since several triplets can encode the
same amino acids.
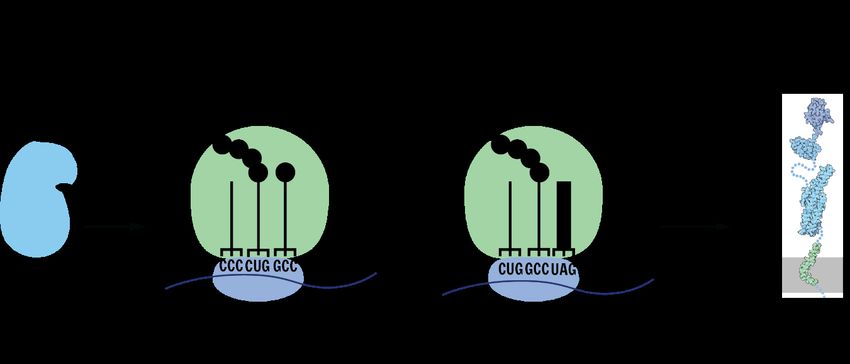
To catalyse eukaryotic protein biosynthesis a ribosome consisting of a small (40S) and a large
(60S) subunit is needed. The small subunit binds the mRNA at the 5’-end in a preinitiation
complex with initiator methionyl-tRNA and scans the mRNA until it arrives at the first AUG
codon, which is the start codon. Through GTP hydrolysis, the large subunit of the ribosome
joins, and the initiation factors are released. The large subunit of the ribosome contains three
binding sites. The A-site binds aminoacyl-tRNA, the P-site binds peptidyl-tRNA and the E-site
binds the tRNA before it exits the ribosome. Furthermore, the large subunit contains the
peptidyl transferase centre where the catalysis of peptide bonds occurs. When the start codon
AUG matches with the initiator methionyl-tRNA at the P-site the translation starts. After that
the elongation factor 1α loads the next aminoacyl-tRNA into the A-site of the ribosome. The
ribosomal peptidyl transferase catalyses the peptide bond formation, before the mRNA:tRNA
complex moves three nucleotides further to make place for the next tRNA on the A-site. To
3make sure, that the right tRNA binds a proofreading step is performed by the elongation factor 1α, before GTP is hydrolyzed to move to the next triplet. Eukaryotic release factor 1 (eRF1) recognizes all three stop codons. After eRF1 binding to the A-site, hydrolysis leads to translation termination and the dissociation of the ribosome3. Figure 1: Translation. The mRNA is used as a template to synthesise new proteins. When the ribosome arrives at a stop codon, the translation stops and the peptide chain is released. 2.1.2 Natural expansion of the genetic code Francis Crick proposed the “frozen accident” theory in 1968 to explain why all domains of life use the same 20 amino acids4. He thought that the evolution of the code was stopped when a certain level of cellular complexity was reached, because it would disrupt the whole proteome of the organism 5. However, now over 50 years later, over 500 amino acids used in nature are known. Bacteria and Fungi use non-ribosomal peptides, which are made of amino acids and are connected through synthetases by peptide bonds 6. These nonribosomal peptide synthetases are enzymatic complexes, that do not use DNA as template. The non-ribosomal peptides are extremely diverse in their structure and function 7. Non-ribosomal peptides can be toxins but are also used as antibiotics and cytostatics 8. Not only non-ribosomal peptides where found, but also two extra proteinogenic amino acids. Proteinogenic means that they are incorporated into proteins during the process of translation. 20 of the amino acids are canonical, as described in chapter 2.1.1. The two extra proteinogenic amino acids are called Selenocysteine (Sec) and Pyrrolysine (Pyl). Both are not considered canonical amino acids since they are not assigned to a unique sense codon. Instead, they are encoded by one of the stop codons. 4
2.1.2.1 Selenocysteine
Seleoncystein is considered the 21st proteinogenic amino acid in bacteria, archae and
eukaryotes. Sec is similar to the canonical amino acid cysteine, except that in Sec a selenium
is at the position of the sulphur. The nucleotide triplet that encodes for Sec is the opal stop
codon (UAG). To specify when an opal stop codon should signal the incorporation of Sec, a
Sec insertion sequence (SECIS) is used. The SECIS encodes a stem-loop structure of about
200 nucleotides in the 3’ untranslated segment of the mRNA, which guides Sec incorporation
via Sec-specific translation elongation factors. The finding that Sec is incorporated at a stop
codon was first reported in formate dehydrogenase of Escherichia coli (E. coli) and glutathione
peroxidase of mice 9,10. In humans, 25 genes have been identified that code for enzymes like
peroxidases and reductases that use Sec. Sec is used in active sites of these enzymes since it
shows higher reactivity and a lower pKa. Therefore, these enzymes have up to 100-fold higher
catalytic efficiency than the enzymes with a cysteine in the catalytic site 11.
2.1.2.2 Pyrrolysine
Pyl is a lysine derivate, with a pyrrolyine-ring amidated to the ε-amine of lysine. It is not as
widely distributed in evolution as Sec. To date, Pyl has only been found in methanogenic archea
and bacteria but is absent in all eukaryotes. In the Methanosarcinaceae family of archea, Pyl is
incorporated in enzymes that catalyse the formation of methane from methylamines like the
monomethylamine methyltransferase 12,13.
Pyl is incorporated at amber stop codons by a cognate pyrrolysyl-tRNA synthetase (PylRS)/
pyrrolysyl-tRNAPyl pair unique to Pyl-containing organisms. tRNAPyl (PylT) recognizes amber
stop codons (UAG). The amino acid itself is synthesized by enzymes encoded in the pylBCD
genes 14. In comparison to Sec, it is not clear how the cell distinguishes if the amino acid should
get incorporated at the amber stop codon or not. A Pyl insertion sequence akin the SECIS was
initially postulated, however no special mRNA element has been identified to date for the
incorporation of Pyl 15–17.
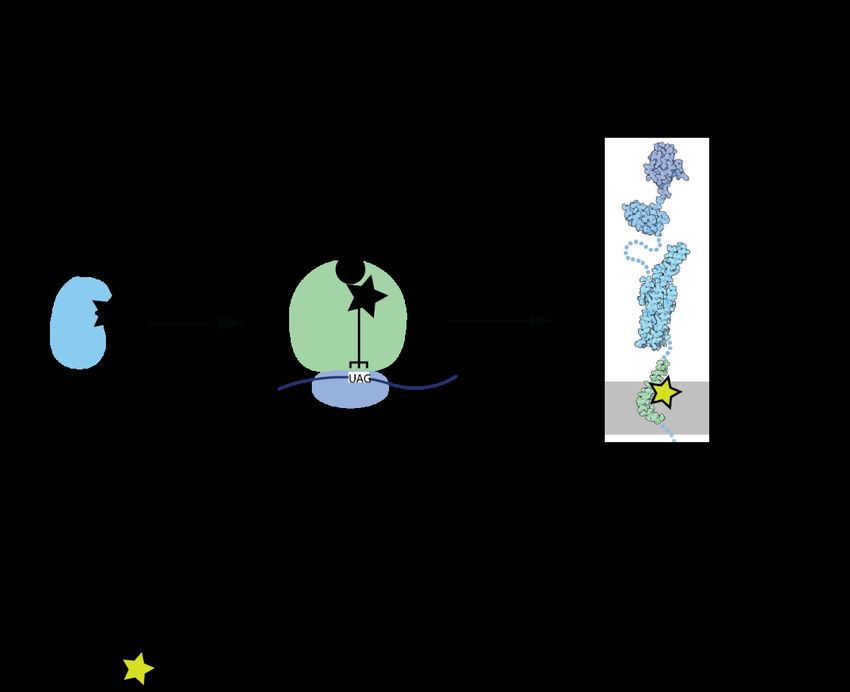
2.1.3 Amber codon suppression
Three components are required to expand the genetic code: 1) A codon to signal incorporation
of the ncAA. The universal genetic code has no blanks since, as described above, every codon
is assigned. Thus, an existing codon must be repurposed for signalling ncAA incorporation,
most often the amber (UAG) stop codon is used. UAG is the least used stop codon in most
organisms, including mammals. Moreover, natural amber suppressors, i.e. tRNAs that suppress
5the use of UAG as stop codon and reassign to another amino acid, have evolved in archea and bacteria (https://www.genscript.com/tools/codon-frequency-table). 2) An orthogonal tRNA-synthetase/tRNA pair. This means, that these biomolecules are not interacting with the endogenous complement of biomolecules in the cells. If orthogonal, the tRNA is not aminoacetylated by any of the endogenous synthetases, and the orthogonal synthetase does not bind and charge any of the endogenous tRNA 18. The orthogonal tRNA recognizes the codon to be suppressed/recoded, while the synthetase charges the orthogonal tRNA with the ncAA. 3) A ncAA. Over 160 ncAAs with a variety of chemical structures can be incorporated into proteins from bacteria, yeast, and mammalian cells. The ncAAs are structurally diverse, but most of them are incorporated by just a handful of different synthetases. The mRNA specifies the order in which the ribosome links the amino acids together. As soon as it reaches the amber codon, normally a release factor binds (eRF1 in eukaryotes), and the peptide chain is released from the ribosome. If an amber suppressor is present, the ncAA is incorporated and the translation continues until the next stop codon (Figure 3). Figure 2: Incorporation of ncAAs via orthogonal synthetase/tRNA pair enables site-specific labelling of proteins 6
2.1.3.1 The orthogonal synthetase/tRNA pair
Several synthetase/tRNA pairs have been found to be orthogonal in bacteria and eukaryotic
cells, but one of the most used orthogonal synthetase/tRNA pairs are the pyrrolysine-
synthetase/tRNAPyl from Methanosarcina barkeri or Methanosarcina mazei (see 2.1.2) 19–21.
The reason for that is the low selectivity of the pyrrolysine-synthetase (PylRS) towards its
amino acid substrate and its orthogonality not only in bacteria, but also in yeast and eukaryotes
22
. Key structural feature recognized by PylRS is that the Pyl amide carbonyl group is separated
from the α-carboxylate by six atoms. Since no canonical amino acid has this structural element,
the synthetase does not recognize any of the canonical amino acids including lysine 23. At the
same time, the minimal recognition features allow PylRS to accept a large range of ε-N-lysine
derivates. Research was done on engineering the synthetase to broaden the possible ncAAs that
can be used as substrate. Yanagisawa et al. mutated two amino acids in the synthetase (Y306A
and Y384F) to incorporate larger lysine derivates, the mutant is hereafter referred to PylRSAF
24
.
2.1.3.2 Non-canonical amino acids
NcAAs can have various functional properties that are useful for biological and life science
applications by containing photocaged groups, biophysical probes, or posttranslational
modifications to control or analyze protein structure. Furthermore, ncAAs can have groups
with chemoselective reactivity 25. This can be used for reacting (hereafter called ‘labelling’)
the incorporated ncAAs with chromogenic compounds and, thus, enable visualization of
26
proteins using microscopy with minimal changes in the proteins structure and function .
NcAA need to be taken up by the cell, metabolically stable, and tolerated by universal
elongation factors of the ribosome 27. Over 200 ncAAs can be incorporated by the orthogonal
28
sythetase/tRNA pairs .
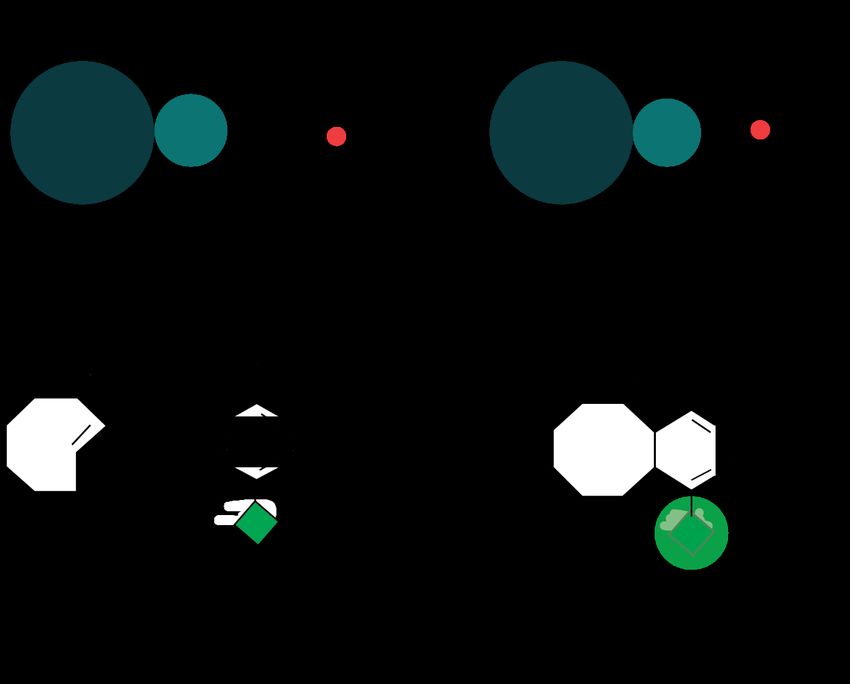
NcAAs, that react via click chemistry reaction with tetrazine dyes are e.g., Cyclopropene-L-
Lysine (CpK), Bicyclo[6.1.0]nonyne–Lysine (BCNK) and Cyclooct–2–en–L–Lysine (TCOK)
(Figure 3). CpK can be incorporated into proteins by wildtypes PylRS/PylT systems, while
BCNK and TCOK require the engineered PylRSAF 29. BCNK has been shown to react with
30
azido- and tetrazine-dyes . Plass et al. synthetized cyclooct-4-ene TCOK and showed its
31
biorthogonality in mammalian cells . However, trans-cyclooctenes were shown to be
accepted by the PylRSAF around three times better than cyclooct-4-ene TCOK 32.
7Figure 3: NcAAs Cyclopropene- L- Lysine (CpK), Bicyclo[6.1.0] nonyne - Lysine (BCNK) and
Cyclooct - 2 - en - L - Lysine (TCOK).
2.1.4 Advantages of amber codon suppression
With GCE, we add a new tool into our toolbox to study molecular mechanisms in neurons. It
is difficult to follow processing products of proteins with traditional labeling methods in living
cells. Fluorescent proteins are used to label the C- or N-terminus of proteins and antibodies can
only be used in fixed cells. Therefore, GCE is a powerful method since proteins can be labeled
on any specific position in the protein.
Furthermore, through the only small change that we make in the protein, it can be also a useful
tool to study peptides 33. Single-residue terminal labeling (STELLA) tags, where a single ncAA
is added to the C- or N-terminus of the peptide, can be used for live-cell imaging 34.
Another area where GCE can be useful is in super-resolution microscopy since the resolution
of the microscopes puts a high demand on the size of the labels. The combination of a primary
and a secondary antibody has a length of up to 30 nm, while super resolution microscopes have
a resolution of 2-40 nm 35. It is even possible to dual-label proteins with GCE in mammalian
32
cells for super-resolution microscopy . The incorporation of different ncAAs can also be
useful in other applications like developing biomaterials or antibody drug conjugates 36,37.
82.1.5 Limitations of amber codon suppression
While GCE offers many advantages, it is also accompanied by methodological limitations.
Through method development, many of the drawbacks like inefficient incorporation of the
ncAA, incorporation of ncAAs into endogenous amber codons and insufficient gene delivery
into the cells were accessed and improved in the last years.
2.1.5.1 Inefficient incorporation of ncAAs
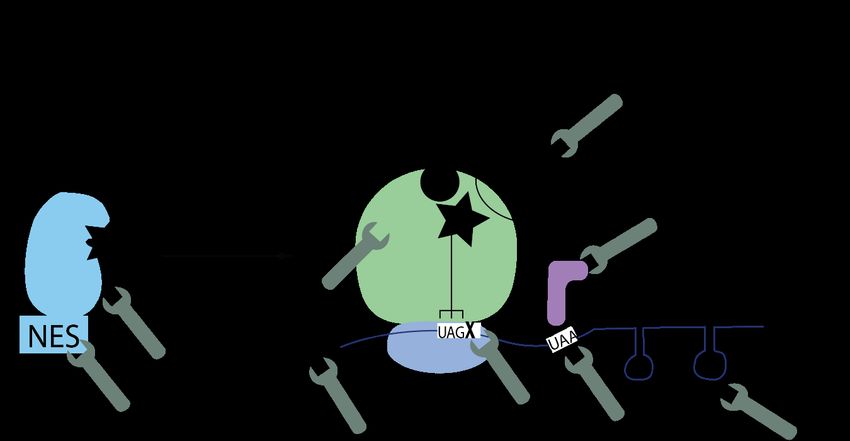
The orthogonal tRNA is always in competition with the release factor when the amber codon
is presented in the A-site of the ribosome. Therefore, the incorporation efficiency can vary
between 10% and 100% 38. However, it is not clear yet what determines the efficiency of the
incorporation. We used the orthogonal PylRS/tRNAPyl pair, which can incorporate Pyl in the
methanosarcinaceae family. As I mentioned in section 2.1.2 no signal is known yet that
determines the incorporation of Pyl into the protein. However, some research showed, that the
4th nucleotide following the amber codon has an influence on the efficiency in E. coli (Figure
4A) 39–41, and a sequence context dependency has been recently studied in mammalian cells 42.
Another way to increase the incorporation of the ncAA is to modify cellular components that
are important in the translation. Such modifications have been explored in E. coli by altering
the properties of for instance the release factor, the ribosome or the elongation factor. 43,44. The
deletion of the release factor one lead to an increase in the incorporation of the ncAA at the
amber codon in E. coli (Figure 4B). As a consequence, some of the endogenous amber codons
45
where mutated to other stop codons to decrease the lethality or all amber codons where
exchanged (Figure 4C) 46,47. Schmied et al. engineered the release factor in mammalian cells to
increase the ncAA incorporation 17-20-fold without increasing readthrough of other stop
codons 29.
Other groups designed new orthogonal ribosome/ mRNA pairs in E. coli, that enabled the
translation of the orthogonal mRNA, whereas endogenous mRNA got translated by
endogenous ribosomes. To achieve that, the Shine-Dalgarno sequence of the mRNA was
changed so that the orthogonal ribosomes, but not the endogenous ones, recognize the mRNA
48,49
(Figure 4 D) . This enables translation of the proteins without competition between the
release factor and the ncAA and can be even used for enabling quadruplet codons for the
incorporation of the ncAA. Quadruplet codons will be discussed in the next section.
The translation elongation factors are not only responsible for the translocation of the amino
acetylated tRNA complexes to the ribosome, but also for proofreading if the tRNAs are loaded
with the correct amino acids 50. Therefore, the incorporation of the orthogonal tRNAs can be
9inefficient since the thermodynamic interaction between the elongation factor and the amino
acetylated tRNA is not correct. Groups engineered the elongation factor either by mutating
them to have a broader substrate acceptance or by engineering novel elongation factors that
are, in addition to the native elongation factor, added to the cells (Figure 4E) 51,52.
The ratio between the orthogonal synthetase and the tRNA plays another important role in the
efficiency of the incorporation of the ncAAs. It was shown that an increased delivery of tRNAs
into the cells increases the suppression efficiency. Therefore, strong RNA polymerase III
29,53–55
promotors like U6, H1 and 7SK are regularly used . Also engineered tRNAs with
mutations e.g., in the hinge between the D- and T-loop can improve the incorporation efficiency
56,57
. However, an increased number of tRNAs can lead to more background labelling, which I
discuss in section 2.2.2 (Figure 4F) 58.
Another approach could be to bring the components that are needed for GCE closer together in
the cells. One way could be to add a nuclear export signal to the synthetase. Using this
approach, an increase in the incorporation efficiency of up to 15 times was shown 59,60, while
other research showed that different nuclear export signals can have different effects on the
ncAA incorporation efficiency even though the localisation is changed from nuclear to
cytosolic 61 (Figure 4G). Another option is to bring the components for GCE in membraneless
organelles together. Most organelles have membranes, but some like nucleoli do not have a
limiting membrane. These membraneless organelles concentrate proteins and RNAs to enhance
reaction efficiency between the accumulated compounds 62. Reinkemeier et al. showed that
these membraneless organelles with the mRNA and the tRNA/PylRS pair for GCE can be
generated. Therefore, the ms2 stem-loop structure from the phage genome, which binds to a
phage-derived major capsid protein, was added to the mRNA and the protein to the PylRS.
This increased the incorporation efficiency up to eight times (Figure 4H) 63,64.
10Figure 4: Engineering cellular components to improve the amber codon suppression efficiency
Wrench A) Optimizing the mRNA context of the amber codon B) Engineering the release factor
to not recognize the amber codon C) Mutating all endogenous amber codons to other stop
codons D) Expressing orthogonal ribosomes, that only recognize mRNA with engineered
sequence E) Optimizing the elongation factor F) Engineered tRNAs to improve the
incorporation efficiency G) Nuclear export signal (NES) improves the incorporation efficiency
of the ncAA H) Membraneless organelles bring all components for GCE in close proximity
2.1.5.2 Incorporation of ncAA into endogenous amber codons
The incorporation of ncAAs into endogenous amber codons can have toxic effects and can lead
to background labeling of other proteins with endogenous amber codons. However, the cells
have a protein quality control, which recognizes incorrectly folded or elongated proteins and
65
degrades them efficiently . Different research groups assessed this problem in GCE by
developing organisms without endogenous amber stop codons or by incorporating ncAA not
into the amber codon, but in quadruplet codons or even in codons with unnatural nucleotides
66 46,67,68
. Several groups removed all amber stop codons in E. coli or even yeast , but his
approach has limitations, since not all mutations are tolerated well and only one ncAA can be
incorporated into the protein 66.
Therefore, quadruplet codons could help to expand the genetic code. Theoretically 256
quadruplet codons could be used for the incorporation of ncAAs with tRNAs that have four
nucleotides in their anticodon loops. However, the incorporation efficiency with the
endogenous ribosomes is very low. Neumann et al. designed an orthogonal ribosome (ribo-Q1)
to decode several quadruplets and the amber codon 69. Schultz and his group even developed
11tRNAs to use codons with five nucleotides, which would increase the number of possible
ncAAs used at the same time even more 70,71.
A more recent development is the use of unnatural nucleotides as a signal for the incorporation
of ncAAs. This approach is extremely challenging, since the unnatural nucleotides need to be
taken up by the cells, incorporated into the DNA and accepted by the proofreading mechanism.
Afterwards the DNA needs to be transcribed into mRNA and translated to proteins. Promising
unnatural nucleotide pairs are the deoxynucleoside d5SICS/dNaM and dTPT3/dNaM pairs,
which were used in E. coli. Therefore, the amount of different incorporated ncAA is
theoretically unlimited and neither truncated protein nor incorporation of the ncAAs into
endogenous proteins can occur. However, until now only three of the codons were shown to be
orthogonal and encode ncAAs 72–74.
2.1.5.3 Gene delivery into the cells
Efficient gene delivery into cells is important in order to understand the function and track the
subcellular location, processing, and turnover of proteins. Furthermore, to establish GCE in
cells, an efficient transfection or transduction is necessary, since the orthogonal tRNA/tRNA-
synthetase pair and the protein of interest with blank codon must be efficiently expressed.
However, introduction and expression of exogenous proteins is challenging in postmitotic cells
like primary neurons.
One way of transfecting cells is via lipofection. Lipofectamine is a cationic based reagent,
which is used for transfection of a wide range of constructs and can be used with many different
mammalian cells. The cationic lipids form unilamellar liposomes, which consists of a single
75
bilayer of amphiphilic lipids. These react with the anionic nucleic acids and membrane .
Lipofection is a simple and fast procedure with little adverse effects on the morphology and
viability of the cells. However, transfection efficiency is the limiting factor since only 1-5% of
neurons express the construct 76. Even in the best of circumstances an efficiency of more than
30% as not been achieved 77.
A highly efficient method for introducing and expressing proteins in primary neurons is the
transduction via baculoviruses. The large cargo capacity with over 30 kb and the minimal
cytotoxicity makes it a useful system to introduce the amber codon suppression machinery into
cells. Another advantage in comparison to lentiviruses and other viruses is the stability of
repetitive sequences like the tRNA cassette in the double-stranded DNA virus 78,79. Through
expression of the vesicular stomatitis virus G-glycoprotein (VSV-G), the transduction
efficiency in mammalian cells is enhanced. This so called pseudotyping is very safe, because
12VSV-G is expressed under the insect specific polyhedrin promotor and prevents the virus from
spreading 80. In primary neurons the transduction rates are around 80% and the virus has no
influence on the cell viability, if the medium is changed 10-20 hours after virus application 81.
A third way of expanding the genetic code in cells is the generation of stable cell lines 82. This
approach has several advantages in comparison to transiently transfected or transduced cells.
The first advantage is, that all cells of the stable cell line express the GCE machinery and the
protein of interest with the amber codon. Another advantage is that the approach can be used
to study large cell numbers for biochemical analysis and the experiments are more comparable,
since the number of gene copies integrated into the cell is always the same. A third advantage
is that the cells only need to be transfected and selected once in comparison to transient
transfected cells, that need be transfected for every experiment.
To generate stable cell lines, cells need to still divide, therefore it is not possible in primary
cortical neurons. Antibiotic resistance genes can serve as selection marker to select the
transfected cells from the non-transfected cells. The overexpression of the proteins can be
defined by the concentration of added antibiotics. For the integration the PiggyBac mediated
genomic integration can be used 55. Therefore, it needs two components, the transposon, and a
transposase. This DNA transposon can be integrated by the transposase randomly into different
locations at TTAA sites within the genome.
2.2 BIOORTHOGONAL LABELING
2.2.1 SNAP-tag labeling
If a chemical reaction does not interfere or interact with the biological environment in which it
83
takes place, the reaction is defined as bioorthogonal . One bioorthogonal reaction is the
labeling of SNAP-tags with benzylguanosines with only guanine as side product (Figure 5A).
SNAP-tags consist of the human DNA repair protein O6-alkylguanine-DNA alkyltransferase
(hAGT). The tag must be expressed at the C- or N-terminus of the protein. The enzyme
transfers an alkyl group of its reaction partner O6-benzylguanine to one of its cysteine residues
84
. hAGT has a relatively low substrate specificity and accepts also oligonucleotides that
contain derivates of O6-benzylguanosine with substituted benzylrings 85. This made it possible
to develop different SNAP-dyes. However, many of the dyes show either high non-specific
86
labeling, while others photo-bleach rapidly . Through optimization and development of
improved dyes and SNAP-tags it was possible to use the SNAP-tag labeling for different
applications like super-resolution microscopy, analysis of protein functions or in vivo labeling
87–89
of neurons . Even wash-free systems were developed that have a reduced fluorescent
13background through an incorporated quencher on the guanine group 90. The SNAPf is a fast-
labeling variant of the SNAP-tag with a ten times increased reactivity with a benzylguanosine.
Labeling with small organic dyes is preferable to labeling with fluorescent proteins, because of
their stability and brightness.
2.2.2 SPIEDAC reaction
Other bioorthogonal reactions are click-chemistry reactions. Barry Sharpless, the originator of
Click-Chemistry won the Nobel prize in 2001. He and Finn discovered the copper catalyzed
91
azide-alkyne 1,3-dipolar cycloaddition (CuAAC) . This bioorthogonal reaction is fast but
needs copper as catalyst. Therefore, the reaction can be toxic for the cells. Another
disadvantage is that copper can affect the fluorescence of fluorescent proteins like GFP 92. A
catalyst-free reaction is the strain promoted azide-alkyne cycloaddition (SPAAC) reaction.
This cycloaddition between a cyclic alkyne and an organic azide is slower than the CuAAC
93
reaction but takes place at room temperature and in aqueous environment . Another
bioorthogonal reaction, which is faster than SPAAC, is the cycloaddition between a conjugated
diene and a substituted alkene, which is called strain-promoted inverse-electron-demand Diels–
Alder cycloaddition (SPIEDAC) reaction. This reaction is used to label proteins that contain
ncAAs with strained alkenes, like the ncAAs CpK, BCNK and TCOK with tetrazine dyes
(Figure 5B) 94. The cycloadditons are, with rates of 102-104 M-1s-1, extremely fast and nitrogen
gas is the only byproduct 95,96.
Bioorthogonal reactions can be orthogonal to each other. Nikic et al. showed, that with
carefully selected tetrazine-dyes and dienophiles multicolor labeling of live cells is possible 32.
TCOK reacts with H-Tetrazine-dyes and Met-Tetrazine dyes, while the ncAA cyclooctynyl-
lysine derivative only reacts with H-Tetrazine-dyes. This enables dual labeling, depending on
the incorporated ncAA.
14Figure 5: Bioorthogonal labeling A) SNAP-tag labeling with benzylguanine-derived dye B)
SPIEDAC reaction between the ncAA TCO*K (axial isomer) and a tetrazine dye.
2.2.2.1 Tetrazine-dyes
There are more requirements for the tetrazine-dyes, than that they need to react with the ncAAs.
The fluorescent dyes must 1) be bright and photostable, 2) absorb long wavelength light to
cause less phototoxicity, 3) be stable in water and 4) have little background. Rhodamine dyes
like silicon rhodamine-tetrazine fulfill the first three criteria, but are known to accumulate in
mitochondria and produce a high background fluorescence 97,98.
Turn-on dyes like 6-methyl-tetrazine-BODIPY-FL (BDPFL) or Tetrazine-ATTO-488
(ATTO488) increase their fluorescence after binding to ncAAs. The “turn-on” effect is based
on a quenching mechanism between the dyes and the tetrazine group, which stops as soon as
the dye binds the ncAA 99. Thereby the tetrazine group functions as quencher and bioorthogonal
reactant. The quenching occurs through Förster resonance energy transfer (FRET) from the dye
to the tetrazine 100.
15BDPFL has a 15-times increased fluorescence signal, which makes the BDPFL dye suitable
for live cell imaging. Furthermore, the dye is cell permeable so that intracellular targets can be
labeled. However, BDPFL is less suitable for super-resolution microscopy due to its low photo-
stability 101.
ATTO488 has a 5 to 15 times increased fluorescent signal after binding to the ncAA but shows
a decrease in fluorescence intensity over time after binding to TCOK. A reason for that could
be the decarboxylative elimination (see section 2.2.2.2). The turn-on ratio of H-Tet-ATTO488
is three times lower than the one of Me-Tet-ATTO488, but the reaction with TCOK is around
30 times faster 94.
Another cell-permeable dye is Janelia Fluor 549, which is a tetramethylrhodamine analog.
Through exchange of an N,N-dimethyl amino group with an azetidine constitute, the dye is two
times brighter than tetramethylrhodamine in living cells. The cell impermeable dye Tetrazine-
Cy3 is a bright dye that has an excitation maximum around 550 nm. It was shown that the dye
reacts via SPIEDAC reaction with TCOK 102.
2.2.2.2 Limitations of SPIEDAC reactions
Background labeling of tetrazine-dyes is a big challenge in the GCE field. Not only background
labeling through incorporated ncAAs at endogenous amber codons (section 2.1.4) or free dye,
but also background labeling in the nucleus and cytosol from dye that bound to ncAA coupled
to tRNA or synthetase. A possible solution is adding a nucleus export signal to the synthetase
to reduce the background in the nucleus 60. Another solution could be a reduction of tRNAs.
Since dyes that bound to the ncAA coupled to tRNA cannot be washed out, cells, transfected
58
with plasmids just carrying a single copy of PylT, show reduced background labeling .
However, this could also reduce the incorporation efficiency of the ncAA.
Arsic et al. showed background labeling in lysosomes through accumulation of ncAAs and
subsequently binding of the tetrazine-dyes. However, a higher overexpression of the protein of
interest with amber codon seems to reduce the background 103. Also, the reduction of ncAAs
in the cells can improve the signal to noise ratio 98. A second limitation can be the elimination
of the protein from the transcyclooctene after binding to the tetrazine dye, if TCOK is used as
ncAA (Figure 6). This rection is pH sensitive, which means that it could play a role when
proteins are studied, that are found in acidic organelles like lysosomes or endosomes 104. This
reaction can also be used for a prodrug release but is a limitation for GCE 105. However, NMR
measurements showed that trans-TCO*K is more stable than other TCOKs in PBS/dioxane-d8
32
.
16Figure 6: As a second step after the SPIEDAC reaction, a decaging reaction can occur, where
the protein gets eliminated from the tetrazine dye by binding to cyclooctane.
2.3 GENETIC CODE EXPANSION FOR NEURONAL STUDIES
GCE adds a new tool to study biochemical processes in neurodevelopment, neurodegeneration,
and other fields of neuroscience. This tool is used in different organisms like bacteria, insects,
mammalian cells and even mice. In this literature review, I focus on the use of GCE to study
neurological processes in mammalian cells, especially neurons and mice.
2.3.1 Genetic code expansion in cell lines
Ion channels and neurotransmitter receptors are key features of neurons. They control neuronal
communication, synaptic transmission, and the membrane potential. GCE is a popular tool to
study their dynamics with photoswitchable ncAAs to optocontrol their activity. Furthermore,
the influence of specific amino acid positions in the binding domains can be determined with
GCE.
Already in 2007, Wang et al. used GCE to study the Kv1.4 channel, a fast inactive A-type
Potassium channel, which is widely distributed in the central nervous system. They found that
the bulkiness of the amino acid residues determines the fast channel inactivation 53. The first
receptor that was studied with GCE was the α-amino-3-hydroxy-5-methyl-4-
isoxazolepropionic acid (AMPA) receptor, which is responsible for fast excitatory synaptic
transmission. By UV-light the AMPA receptor subunit GluA2 with an incorporated 4-Benzoyl-
106
L-phenylalanine (BzF) was inactivated irreversibly .
The neuropeptide substance P is responsible for physiological responses such as transmission
of pain and inflammation through the Neurokinin-1 receptor. Rannversson et al. mutated 34
positions in this receptor to incorporate the ncAA BzF 107. Thus, they were able to study the
binding sites in the G-protein coupled receptor for substance P. They also used the tool to study
binding sites in the human serotonin transporter for antidepressant drugs like escitalopram and
paroxetine 108,109.
17The first reversible light-switching of ncAAs was performed by Klippenstein et al. They
incorporated a photo-switchable ncAA into an N-methyl-D-aspartate (NMDA) receptor and
stimulated with light the switch of the ncAA configuration, which influenced the conformation
of the receptor 110. The incorporation of the ncAA into an NMDA receptor was also used to
demonstrate the superiority of GCE to conventional immunocytochemistry for quantitative
imaging 111. Furthermore, photoswitchable ncAAs have been used to study other receptors and
channels in the last years 112–115.
The Tyrosine kinase A is a membrane receptor, which could be used as a therapeutic target for
neurologic diseases. Therefore, site-dependent phosphorylation of the receptor was studied
116
with GCE in HEK293 cells and in SH-SY5Y cells . The latter cells have neuronal
characteristics since they are neuroblastoma cells. However, GCE was also established in
primary neurons from mice and rats.
2.3.2 Genetic code expansion in primary neurons
The application of GCE and the subsequent SPIEDAC reaction between Tetrazine-dyes and
ncAAs is extremely challenging in primary neurons. The two main obstacles are the low
transient transfection efficiency of primary neurons and the high background labeling of the
tetrazine dyes. Nevertheless, several groups established GCE in primary neurons. The proof of
principle was shown in 2007 by Wang et al. who transduced mouse hippocampal neurons with
pCL-Ampho Retrovirus expressing a reporter consisting of GFP with an amber codon at
position 182 53.
The first biological relevant protein that was studied with GCE in neurons was the voltage-
dependent membrane lipid phosphatase, a member of voltage-dependent proteins that are not
ion channels. The neural stem cell line HCN-A94 from rat brain were transduced via
lentiviruses with the GCE machinery and the phosphatase. Subsequently the cells were
differentiated to neurons and the fluorescence change of the ncAA 2-amino-3-(5-
(dimethylamino) napththalene-1-sulfonamido) propanoic acid, which is sensitive to
environmental polarity, was measured 117.
To increase the copy number of expressed tRNA, neurons needed to be transduced with viruses
where less homologous recombination events occur than in lentiviruses. Therefore, Chatterjee
et al. developed a baculovirus system to transduce neurons and brain slices with the GCE
machinery and a GFP reporter. However, they detected mainly transduced glial cells and less
than 5% transduced neurons 78,79.
18Another important step towards studying physiological processes in primary neurons was made
by introducing the protein of interest via CRISPR/Cas into the cells. A FLAG-tag was added
on the C-terminus of the neurofilament light chain, a biomarker for many neurodegenerative
diseases. The amber codon was added in the linker between the FLAG-tag and the protein, so
that the endogenous levels of the protein where maintained, even with an incorporation
efficiency of the ncAA of less than 100% 103.
Two AMPA receptor auxiliary subunits of the transmembrane AMPA regulatory protein
family were labeled with GCE in living primary neurons and organotypic brain slice cultures.
To reduce the toxicity of ncAAs that got incorporated into endogenous amber codons, the
PylRS and AMPA subunits where overexpressed under a bidirectional doxycycline inducible
promotor. With this system they were able to image the subunits with super-resolution
microscopy in primary neurons 118.
2.3.3 Genetic code expansion in vivo
61
GCE was also performed in different animals like Caenorhabditis elegans , Drosophila
melanogaster 119 and Mus musculus 120. Using the GCE tool for studying neuronal functions in
living mice is difficult and expensive, since the ncAA needs to penetrate the blood-brain-barrier
after being administered orally in the drinking water or need to be injected intraperitoneally.
Kang et al. incorporated a photoreactive ncAA into the pore of an inwardly rectifying
potassium channel Kir2.1. They electroporated the construct together with the GCE machinery
into mouse embryos in utero and measured the changes in the current through light-activation
in the neurons, showing that they were able to activate the channel through the incorporated
ncAA. 121.
An adeno-associated virus with a reporter consisting of super-folded (sf)GFP with the amber
codon at position 150 and the GCE machinery was stereotactically injected into the region of
the suprachiasmatic nucleus of mouse brains. However, they not only found sfGFP expressing
cells in that nucleus but also in neurons in the hypothalamus surrounding 122. Cryptochromes
play an important role in regulating the suprachiasmatic nucleus, the central pacemaker of the
circadian rhythm. Therefore, adeno-associated viruses carrying Cryptochrome 1 with an amber
codon were injected into the suprachiasmatic nucleus of cryptochrome knock-out mice. Adding
the ncAA into the drinking water of the mice rescued the circadian rhythm of the mice, showing
that the ncAA got incorporated into Cryptochrome 1. 123.
19The first transgenic mice expressing a methionyl-tRNA synthetase to detect newly synthesized
proteins in neurons have been developed 124. The methionyl-tRNA synthetase has an expanded
amino acid binding site, so that it can be charged with the ncAA azidonorleucine. Therefore, it
is possible to detect proteins, which were synthesized at a specific time point. As long as the
ncAA is added, the ncAA will be incorporated into the proteins at methionine codons. With
this method, the scientists were able to detect proteins which expression is regulated by sensory
input 124,125. This bioorthogonal ncAA tagging (BONCAT) method was also used to identify
cell-specific proteomes. To achieve this, the synthetase was expressed under a neuron-specific
human synapsin 1 promoter 126.
2.3.4 Genetic code expansion in human cells
The field of GCE was expanded in the last year to study proteins in human cells. Shao et al.
used self-replicated plasmids, derived from the Epstein-Barr virus to suppress the amber codon
in a GFP reporter in human hematopoietic stem cells 127. They not only tested this one-vector
system in vitro, but also engraved the engineered hematopoietic stem cells into mice. They
were able to detect differentiation of the stem cells into B-cells, myeloid-cells, and T-cells. To
further expand the possible cell types for GCE in human cells, we developed stable hiPSCs
with a GCE machinery and differentiated them to neurons 128. However, these hiPSCs could
also be differentiated in the future to other cell types. This will be discussed in section 5 and 6
of this thesis.
2.3.5 Genetic code expansion to study AD
APP is a key player in AD. It gets processed by three different secretases in neurons, which
leads to many cleavage products. To study the cleavage products from the middle of the protein,
like the neurotoxic Aβ, in living cells, GCE is a suitable method 129. Therefore, we dual-labeled
130
APP with a ncAA in the Aβ region and a tag on the C-terminus . A similar study was
subsequently reported by another group 131. While we used a SNAP-tag to label the C-terminus,
Sappakhaw et al. used the fluorescent protein GFP or a HALO-tag. These experiments where
performed in HEK293T cells. We found that incorporation of the ncAA instead of a histidine
on position 13 in Aβ works successfully and without influencing the Aβ42 production 130. We
were happy to see that this position was also the best working positions in the APP-GFP
construct 131. Sappakhaw et al. labeled the ncAA with the cell impermeable tetrazine-Cy5 dye
to label APP in the cell membrane. They added a quencher on the HALO-tag and followed the
endocytosed labeled APP in the HEK293T cells. Therefore, they were able to detect de-novo
generated Aβ-peptides when the quencher on the C-terminus got released from the Aβ-peptide.
20You can also read

















































