Structural arrangement of the VH and VL domains in the COBRA T-cell engaging single-chain diabody - Oxford Academic
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Antibody Therapeutics, 2022, Vol. 5, No. 1 1–10
https://doi.org/10.1093/abt/tbab028
Advance Access Publication on 16 December 2021
Original Research Article
Structural arrangement of the VH and VL domains in
the COBRA™ T-cell engaging single-chain diabody
Jessica Krakow1 , Michal Hammel2 , Ying Zhu1 , Brian J. Hillier1 , Bryce Paolella1 ,
Austin Desmarais1 , Rusty Wall1 , Tseng-Hui T. Chen1 , Rex Pei1 , Chulani Karunatilake1 ,
Robert DuBridge1 and Maia Vinogradova1 , *
1 Maverick
Therapeutics, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, Brisbane, CA,
USA, and 2 Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA,
Downloaded from https://academic.oup.com/abt/article/5/1/1/6463575 by guest on 28 January 2022
USA
Received: August 12, 2021; Revised: November 19, 2021; Accepted: November 22, 2021
ABSTRACT
Background: COBRA™ (COnditional Bispecific Redirected Activation) T-cell engagers are designed to target
solid tumors as a single polypeptide chain prodrug that becomes activated by proteolysis in the tumor
microenvironment. One COBRA molecule comprises seven Ig domains: three single-domain antibodies
(sdAbs) recognizing a tumor target or human serum albumin (HSA), and CD3ε-binding variable fragment
heavy chain (VH) and variable fragment light chain (VL) and their inactivated counterparts, VHi and VLi.
Pairing of VH and VL, and VLi and VHi into single-chain variable fragments (Fv) is prevented by shortened
inter-domain linkers. Instead, VH and VL are expected to interact with VLi and VHi, respectively, thus making
a diabody whose binding to CD3ε on the T-cells is impaired.
Methods: We analyzed the structure of an epidermal growth factor receptor (EGFR) COBRA in solution using
negative stain electron microscopy (EM) and small-angle X-ray scattering (SAXS).
Results: We found that this EGFR COBRA forms stable monomers with a very dynamic interdomain
arrangement. At most, only five domains at a time appeared ordered, and only one VH-VL pair was found
in the Fv orientation. Nonenzymatic posttranslational modifications suggest that the CDR3 loops in the VL-
VHi pair are exposed but are buried in the VH-VLi pair. The MMP9 cleavage rate of the prodrug when bound
to recombinant EGFR or HSA is not affected, indicating positioning of the MMP9-cleavable linker away from
the EGFR and HSA binding sites.
Conclusion: Here, we propose a model for EGFR COBRA where VH and VLi form an Fv, and VL and VHi do
not, possibly interacting with other Ig domains. SAXS and MMP9 cleavage analyses suggest that all COBRA
molecules tested have a similar structural architecture.
Statement of significance: The design of the COBRA platform utilizes a combination of various
immunoglobulin domains, including single domain antibodies and variable fragment heavy chain (VH)
and variable fragment light chain (VL) domains of antibody Fab regions that are expected to form a
diabody. Our study showed that the structural arrangement of the domains in COBRA is different from
the expected yet the stable fold is maintained. Findings provide insights for design of the novel protein
therapeutics.
KEYWORDS: antibody; protein therapeutic; T-cell engager; diabody; structure; SAXS
∗ To whom correspondence should be addressed. Robert DuBridge or Maia Vinogradova. Email: maia.vinogradova@takeda.com or
bob.dubridge@takeda.com
© The Author(s) 2021. Published by Oxford University Press on behalf of Antibody Therapeutics. All rights reserved. For Permissions,
please email: journals.permissions@oup.com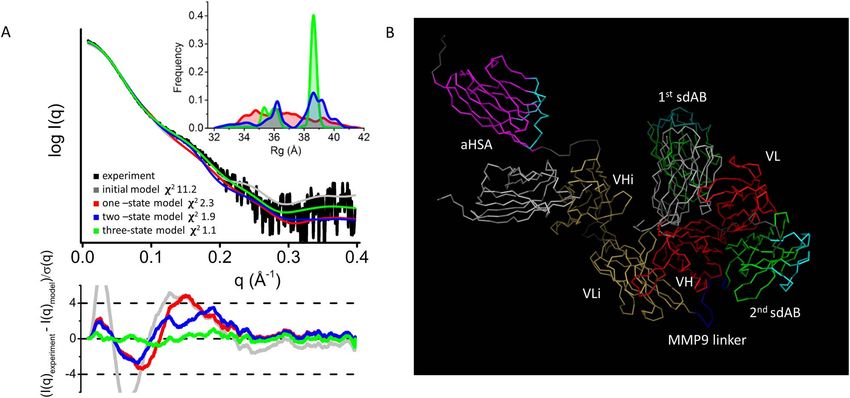
2 Antibody Therapeutics, 2022
INTRODUCTION with each other or with themselves in a manner different
from typical Fv interactions.
Bispecific antibodies targeting two different antigens, The interactions between VH and VL in the Fv inter-
or two different epitopes of the same antigen, are a face of a diabody are dynamic. As revealed in [11], the
growing class of recombinant protein therapeutics [1]. single-chain bispecific diabody featuring VH1-VL2–42 aa
Design of bispecific antibodies often utilizes the IgG linker -VH2-VL1 format, where each VH-VL pair was
format of a classical monoclonal antibody, with the Fc separated by five amino acid linkers, showed functional bis-
region contributing to the mechanism of effector function pecific binding when purified. However, after the cleavage
in antibody-dependent cellular cytotoxicity (ADCC), of the 42-amino acid linker with thrombin, a nonfunctional
complement-dependent cytotoxicity (CDC) and molecular monospecific diabody (VH2-VL1)2 has been crystallized.
recycling via binding to FcRn receptors. The variable VH-VL pairs were rearranged under the crystallization
regions of IgG in bispecific antibodies are designed conditions that typically take place at high protein concen-
to have different independent binding functions and trations. The authors argued that the changes in confor-
sometimes are added to, or replaced by, alternative binding mation of the heavy chain CDR3 loop caused this domain
modules such as single-chain variable fragments (scFvs),
Downloaded from https://academic.oup.com/abt/article/5/1/1/6463575 by guest on 28 January 2022
shuffling.
single-domain antibodies (sdAbs), diabodies and fusion Recently, the conditionally active COBRA™ (COndi-
proteins. Bispecific antibodies that do not utilize the tional Bispecific Redirected Activation) T-cell engager has
IgG format include bispecific T-cell engagers (BiTEs) [2], been described [12]. The molecule encodes a single-chain
dual affinity re-targeting antibodies (DARTs) [3–4] and diabody (Fig. 1) in the VH-VL and VLi-VHi format where
tandem diabodies (TandAbs) [5]. Their design utilizes VH and VL are heavy and light chain variable fragments
variable fragment heavy chain (VH) and variable fragment of a CD3ε-binding antibody. VLi and VHi are inactivated
light chain (VL) domains of antibody Fab regions linked versions of these same fragments, with modifications in the
together in one polypeptide chain forming an scFv, or CDR2 loop. VH and VL, as well as VLi and VHi, are linked
placed on two complimentary polypeptide chains forming by short GGGGS linkers and these pairs are separated by
a diabody. Bispecific function of such engineered antibodies an sdAb which binds to the tumor target protein (second
is achieved by connecting two or more VH-VL pairs in the sdAb in Fig. 1). At the N-terminus, the COBRA molecule
final quaternary or tertiary structure. BiTEs have two scFvs has another identical tumor binding sdAb and finally an
linked together and each VH and VL are connected by a anti-HSA sdAb is located at the C-terminus. Due to the
15-aa long (GGGGS)n linker. In DARTs and TandAbs the short linker length between VH-VL and VLi-VHi pairs,
linkers between VH and VL are short, up to 9 amino acids which prevents formation of scFvs, the pairs were expected
long, to prevent interaction of VH and VL located on the to interact with each other. This would render the intact
same polypeptide chain. Each design has its advantages molecule unable to bind CD3 on T-cells since the antigen-
and disadvantages. Proteins containing scFvs (BiTEs) binding site is destroyed by the CDR2 loop modifications in
showed a tendency for oligomerization and aggregation VLi and VHi. Conditional activation of the molecule takes
that impeded their manufacturing. This problem was place upon proteolysis of the MMP9/2 cleavable linker
partially addressed by DART and TandAbs formats. The positioned between the second sdAb in the middle of the
structures of diabody assembly in DARTs and TandAbs polypeptide chain and the VLi domain. Proteolytically-
and the placement of antigen-binding sites varies, showing produced fragments are free to rearrange and form active
flexibility in the structural organization which is difficult and inactive diabodies. The active VH-VL diabody, while
to predict. still associated with the surface of the tumor cell via sdAb
There are a number crystal structures of diabodies in the domains, can engage T-cells via binding to CD3.
protein databank (PDB). All structures show the expected To understand the interdomain interactions in COBRA
VH-VL orientation, generally as in an Fv, although the molecules, we studied the domain organization of EGFR
geometry of the interface has a certain degree of freedom COBRA in solution by structural and biochemical meth-
[6]. The typical interface of the interacting VH-VL engages ods. While high-resolution structural techniques such as X-
some framework residues and some residues of their CDR ray crystallography and NMR were considered, they were
loops, and CDR3 loops contribute significantly to the inter- not prioritized due to either inability to produce crystals
face [7–9]. However, other additional interactions between of EGFR COBRA in an extensive screening campaign, or
VH and VL are sometimes observed. A diabody with bispe- due to the large size of the molecule. Instead, the struc-
cific function was first described in 1994 [10] and featured a tural information was obtained using low-resolution tech-
VHA-VLB and VHB-VLA format. The crystal structure of niques: negative stain electron microscopy (EM) and small-
a diabody with that design (1LMK.pdb) revealed a dimer angle X-ray scattering (SAXS). We found that although
with VHA-VLA and VHB-VLB Fv pairings as expected. the domain arrangement in EGFR COBRA supports the
Interestingly, the asymmetric unit of the crystal contained anticipated mode of action, it does not rely on diabody
two dimers that were associated with each other via an formation. Folding of EGFR COBRA depends on inter-
extensive interface between heavy chains VHA and VHB actions between VH and VLi via the Fv interface, while
and light chains VLA and VLB. The crystal structure of other domains may or may not interact with each other
a different diabody (6KR0.pdb) also revealed a dimer of without engaging in Fv interactions. Regardless of the
the diabody in the crystal asymmetric unit with the dimer missing second Fv pair formation, EGFR COBRA is sta-
interface formed between the identical Fv surfaces. These ble, monomeric and activatable. It maintains an inactive
examples show that the VH and VL domains can interact conformation prior to MMP9/2 cleavage and demonstrates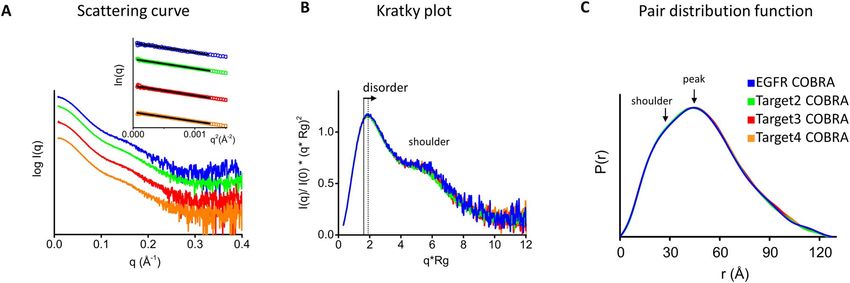
Antibody Therapeutics, 2022 3
Figure 1. Schematics of COBRA domain arrangement in the polypeptide sequence. In green, tumor target binding domains; in magenta, HSA binding
sdAb; in red, heavy and light chains of T-cell CD3ε binding Fv; in yellow, inactivated heavy and light chains of T-cell CD3ε binding Fv. The length of the
linkers and MMP9 cleavage site are indicated.
bispecific T-cell engager activity after cleavage. Analysis was collected at the SIBLYS beamline (BL 12.3.1) at
of several COBRAs with different tumor targeting sdAbs the Advanced Light Source (ALS) at Lawrence Berkeley
showed that all tested proteins had similar structures. National Laboratory (LBNL) in Berkeley, California [15].
Downloaded from https://academic.oup.com/abt/article/5/1/1/6463575 by guest on 28 January 2022
X-ray wavelength was set at λ = 1.216 Å, and the sample-
to-detector distance was 2070 mm, resulting in scattering
MATERIALS AND METHODS vectors (q) ranging from 0.01 to 0.4 Å−1 . The scattering
vector is defined as q = 4π sinθ /λ, where 2θ is the scattering
Materials
angle. All experiments were performed at 20 ◦ C, and data
Human recombinant MMP9 was purchased from R&D were processed as described [16]. Briefly, an SAXS flow
Biosystems (#911-MP-010) and activated according to cell was coupled to an inline Agilent 1290 Infinity HPLC
the manufacturer’s protocol. Recombinant human serum system using an SEC column (Zenix-C, 7.8 × 300 mm,
albumin was purchased from Akron Biotech (Cat#8998- 3 μm particle size, Sepax Technologies Inc, part # 233300–
0001). COBRA molecules were produced recombinantly 7830). The column was equilibrated with mobile phase
in HEK293 cells and purified as described [12]. Tumor (0.1 M sodium phosphate +0.2 M arginine, pH 7.0.) with
proteins targeted by COBRA molecules were produced a 0.45 ml/min flow rate. A total of 50 μl of each sample
recombinantly in HEK293 as His6 - or Fc-tagged fusion was run through the SEC, and 2 s X-ray exposures were
proteins and were purified using Protein A or Ni-NTA collected continuously during a 30 min elution. The SAXS
affinity chromatography followed by SEC. frames recorded before the protein elution peak were
used to subtract baseline noise from all other frames.
The corrected frames were investigated by the radius
Negative staining EM of EGFR COBRA of gyration (Rg) derived by the Guinier approximation
Imaging was done by NanoImaging Services, Inc. (4940 I(q) = I(0) exp(−q2 ∗ Rg∗ 2/3) with the limits q∗ Rg < 1.5.
Carroll Canyon Road, Suite 115 San Diego, CA 92121). The elution peak was mapped by comparing integral ratios
A sample of EGFR COBRA was diluted to 0.01 mg/ml to background and Rg relative to the recorded frame using
with buffer containing 25 mM citric acid, 75 mM NaCl and the program SCÅTTER available at www.bioisis.net. A
75 mM L-Arginine, pH 7.0. The sample was imaged over a homogenous state of COBRA samples characterized by
layer of continuous carbon supported by nitrocellulose on a uniform Rg values across an elution peak was sampled.
400-mesh copper grid (see Supplementary Materials online Final merged SAXS profiles (Fig. 3A and B), derived by
for more details). integrating multiple frames across the elution peak, were
Particles were identified in the high magnification images used for further analysis, including a Guinier plot, which
prior to alignment and classification. Individual particles in determined the sample to be aggregation free (Fig. 3A
the 67 000x high magnification images were selected using inset). Normalized Kratky plots for flexibility assessment
automated picking protocols [13] and manual picking. The were derived from the scattering data. The program SCÅT-
individual sub-images were stacked for processing using TER was used to compute the pair distribution function
reference-free classification based on the XMIPP [14] pro- (P(r)) (Fig. 3C). The distance r where P(r) approaches
cessing package. An initial round of alignments was done zero intensity identifies the macromolecule’s maximal
on each sample and from that alignment class averages that dimension (Dmax, Fig. 3C, Table 1). P(r) functions were
appeared to contain recognizable particles were selected for normalized at the maxima shown in Figure 3C. The SAXS
additional rounds of alignment (Fig. 2). flow-cell was also connected inline to a 1290 series UV–
vis diode array detector (DAD) measuring at 280 and
260 nm (Agilent), 18-angle DAWN HELEOS II MALS and
Size exclusion chromatography coupled to small angle quasi-elastic light scattering (QELS) detectors and Optilab
X-ray scattering with multi-angle light scattering rEX refractometer (all from Wyatt Technology). System
For size exclusion chromatography coupled to small angle normalization and calibration were performed with bovine
X-ray scattering with multi-angle light scattering (SEC- serum albumin using a 45 μl sample at 10 mg/ml in the
MALS-SAXS) experiments (Supplementary Fig. S1), 60 μl same SEC mobile phase and a dn/dc value of 0.175. The
of COBRA samples concentrated to at least 4 mg/ml light scattering experiments were used to perform analytical
were prepared in 25 mM Citric acid, 75 mM L-Arginine, scale chromatographic separations for molecular weight
75 mM NaCl, 4% sucrose, pH 7.0. SEC-MALS-SAXS (MW) determination (Table 1). UV, MALS and differential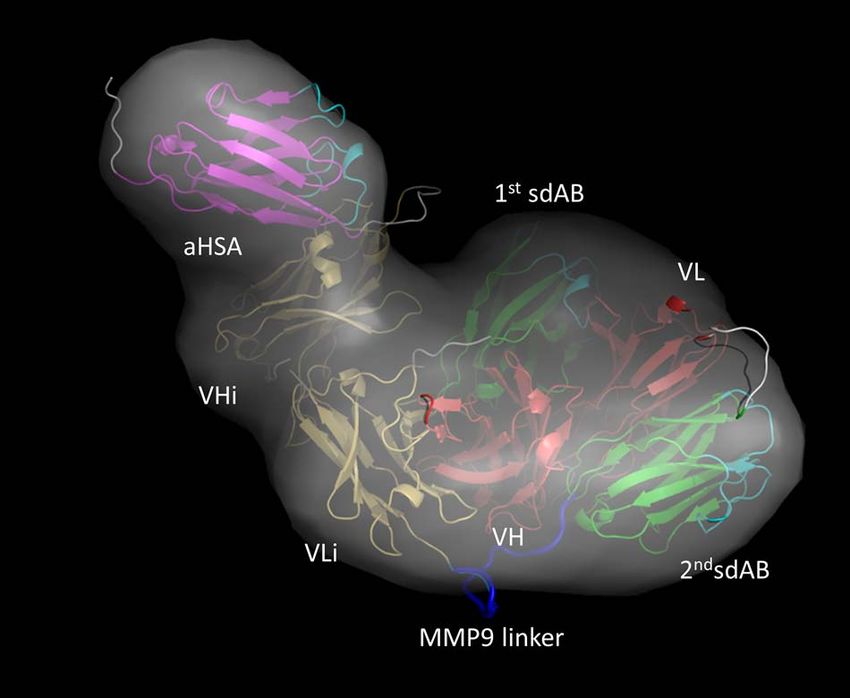
4 Antibody Therapeutics, 2022
Downloaded from https://academic.oup.com/abt/article/5/1/1/6463575 by guest on 28 January 2022
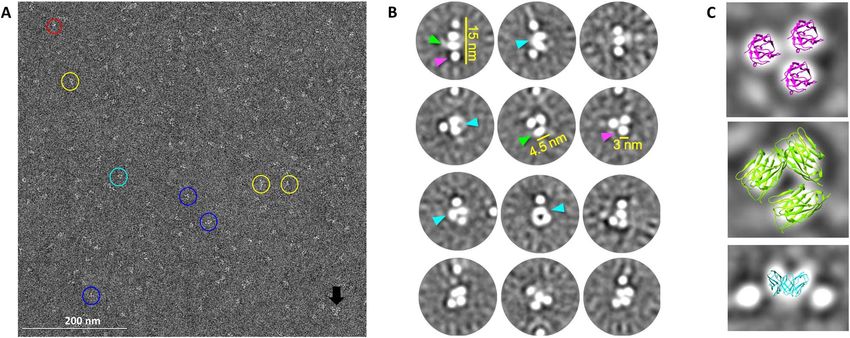
Figure 2. Electron microscopy imaging of negatively stained EGFR COBRA. A- Selected image of EGFR COBRAs at a magnification of 67 000x.
Observed in the sample are: elongated particles (yellow circles); clusters of domains (blue circles); very small particles (red circle); and clumps of particles
(black arrow). In some instances, two oblong shaped domains contact at the base (cyan circle). Scale Bar: 200 nm. B- 2D class averages. The individual
domains are either round (magenta arrows) or oblong (green arrows) in nature, with round domains having a diameter of ∼3 nm and oblong domains
having a longest dimension of ∼4.5 nm. Occasionally two oblong domains come together at one end forming a “V” shape (cyan arrows). C- Overlay of
crystal structures on selected class averages. A random sdAb structure (5VLV.pdb) is shown in two orientations. A “top view” is shown in magenta and
has been fit into circular domains in one of the class averages. A “side view” is shown in green has been fit into oval domains in another class average. The
Fv portion of an anti-CD3ε antibody based on 3R08.pdb is shown in cyan docked into a “V”-shape in a third class average. All images are to scale.
Figure 3. Processed SAXS data for multiple COBRAs. A-Experimental SAXS curves of all tested COBRA samples (colored as shown in panel C). Inset—
Guinier plot with q∗ Rg < 1.5 limit. B- Normalized Kratky plots show similar fold and disorder for all COBRAs (colored as shown in panel C). C- P(r)
functions for all COBRAs normalized onto their maxima. The shift of the main peak toward r ∼ 50 Å indicates the hollowness of particle. P(r) shoulder
at ∼30 Å corresponds to the average size across Ig domains.
refractive index data were analyzed using Wyatt Astra 7 convolution utility [19]. An atomistic model of EGFR
software to monitor sample homogeneity across the elution COBRA was built based on the SAXS envelopes derived
peak complementary to the above-mentioned SEC-SAXS from SAXS data and homology modeling of individual Ig
signal validation. domains (Fig. 4). PYMOL software (The PyMOL Molec-
ular Graphics System, Version 2.0 Schrödinger, LLC) was
used for visualization of the ab initio molecule envelope.
Solution state modeling of EGFR COBRA
The coordinates for the EGFR binding sdAb were from
The ab initio SAXS envelopes were reconstructed from the 4KRL.pdb of 7D12 nanobody with the same sequence in
experimental data on EGFR COBRA using the program complex with EGFR. Homology models of VH, VL, VHi,
DAMMIF [17]. Ten-bead models obtained for each SAXS VLi and HSA binding sdAb were built using the SWISS-
experiment were averaged by DAMAVER [18] to construct MODEL web-based tool of the EXPASY package [20]. The
the average model representing each reconstruction’s models of individual domains were moved manually to fill
general structural features. Bead models were converted the shape of 3D envelope in PYMOL. For modeling of Fv-
to volumetric SITUS formats with the pdb2vol kernel like VH-VL association, alignment with similar structural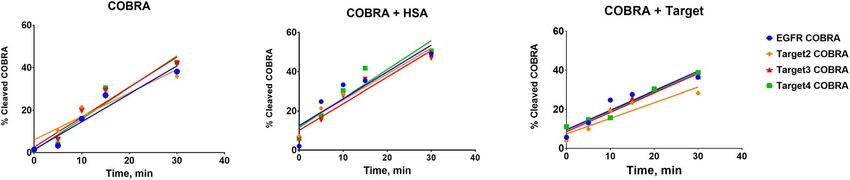
Antibody Therapeutics, 2022 5
Table 1. Physical properties of COBRAs determined by SAXS and MALS
Sample Rg from Guinier plot Rg From P(r) Dmax (Å) MW from Sequence MW from MALS/SAXS
(Å) function (Å) (kDa) (kDa)
EGFR COBRA 37.8 ± 0.6 38.7 ∼125 95 101/91
Target 2 COBRA 37.7 ± 0.2 38.9 ∼125 95 95/94
Target 3 COBRA 38.2 ± 0.4 38.9 ∼125 95 97/94
Target 4 COBRA 37.9 ± 0.6 38.8 ∼125 96 89/94
stored frozen at −20 ◦ C to be analyzed together. SDS-
PAGE was done using pre-cast 10–20% Tris-glycine gels
Downloaded from https://academic.oup.com/abt/article/5/1/1/6463575 by guest on 28 January 2022
(Novex) in Tris-glycine running buffer. Gels were stained
using Instant Blue dye (Abcam) and imaged on an Azure
Biosystems instrument. ImageLab software (BioRad) was
used for quantitative assessment of the protein density in
the stained bands. CE-SDS was performed using a Maurice
instrument (Protein Simple) and quantification was done
using Compass for iCE software.
To assess the MMP9 cleavage of COBRA in the presence
of HSA or tumor target protein, the reaction mixture was
supplemented with either 3 μM of HSA or 4–6 μM of target
protein.
Chymotryptic peptide mapping method
The peptide map of EGFR COBRA was generated by PPD
Inc using the procedure described here. See Supplementary
Figure 4. Modeling of EGFR COBRA. Domains are shown as 3D cartoon Materials online for more details.
models and colored according to the color scheme in Figure 1. CDR loops
in sdAbs are highlighted in cyan. SAXS envelope of EGFR COBRA is
shown as surface in grey.
RESULTS
domains in the crystal structure of diabody 5FCS.pdb Negative stain EM reveals flexible interdomain
was performed. Finally, the linkers between domains were organization in EGFR COBRA
built from the EGFR COBRA polypeptide sequence using To understand the structure of EGFR COBRA, we applied
the program MODELLER [21]. BILBOMD [22] rigid- negative stain EM followed by single particle analysis that
body modeling was applied to the aHSA-VHi and the N- allowed us to visualize domains in this molecule at low
terminal first sdAb linkers to explore the conformational resolution.
space of aHSA and first sdAb domains relative to the EGFR COBRA in solution is typically a monomer
EGFR COBRA core region (Fig. 5). This conformational (>97.5% by analytical SEC) with MW around 93 kDa as
sampling along with an FoXS and MultiFOXS [23–24] determined by SEC-MALS, close to the predicted 95 kDa
approach was used to define, select and weight the one MW [12]. We subjected a sample of EGFR COBRA of
and two-state models that best agree with individual SAXS similar quality, with very little aggregation observed by
profiles (Fig. 5). SEC-MALS, to EM imaging. EM images showed that the
protein particles of EGFR COBRA were well-dispersed,
with only a few instances of clumping observed, confirming
Assessment of MMP9 cleavage of COBRA in vitro using
little aggregation in the sample (Fig. 2A).
SDS-PAGE and CE-SDS
Visible particles showed variation in size and mor-
For cleavage reactions, COBRA at 2 μM was added to phology (Fig. 2A). The elongated particles varied from
activated MMP9 at 2 nM. The reaction was carried out ∼ 11 to ∼ 16 nm in length and from ∼ 2.3 to ∼ 6 nm
in pH 7.0 buffer containing 25 mM citric acid, 75 mM in width. These particles contained distinct individual
NaCl, 75 mM L-arginine, 4% sucrose and 10 mM CaCl2 domains ranging from ∼ 2.3 to ∼ 5.5 nm in their longest
at room temperature. Aliquots of reaction mixture were dimension arranged in a row. Other type of particles
taken out at different time points and mixed with either contained domains of the same size assembled into
SDS-PAGE loading buffer or CE-SDS sample buffer under clusters. Within these rows and clusters, two domains were
nonreducing conditions. Samples were prepared for sub- occasionally observed contacting each other (see cyan circle
sequent analysis by heating at 80 ◦ C for 2 min and were in Fig. 2A).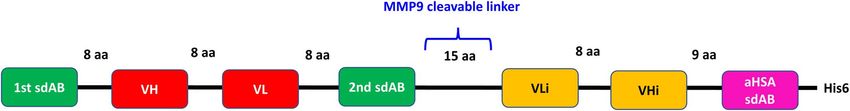
6 Antibody Therapeutics, 2022
Downloaded from https://academic.oup.com/abt/article/5/1/1/6463575 by guest on 28 January 2022
Figure 5. Multistate model of EGFR COBRA. A- Experimental SAXS profile of EGFR COBRA (black) and theoretical SAXS profiles calculated from
their respective initial, one, two and three-state model (gray, red, blue and green) are shown together with residuals (Experiment/Model). The significant
improvement in the SAXS fit for the multistate model indicates that the EGFR COBRA flexibility is required to match the experimental SAXS curve.
B- Domains are shown as ribbon models and colored according to the color scheme in Figure 1. CDR loops in sdAbs are highlighted in cyan. The initial
positions of the first sdAb and aHSA sdAb are shown in grey.
The 2D class averages obtained for EGFR COBRA SEC-MALS-SAXS analysis of EGFR COBRA
(Fig. 2B) were well-defined and consistent with initial imag-
To further assess the flexibility of the EGFR COBRA
ing observations. One class of 2D averages showed that
molecule and to look at its structure using an alternative
elongated particles were composed of three to four well-
low-resolution biophysical technique, we measured small
defined individual domains stacked in a row. Other classes
angle X-ray scattering from the EGFR COBRA protein
of particles appeared as clusters of one to five individ-
particles in solution.
ual domains. The domains were either round or slightly
The SAXS sample cell was connected to an analyt-
oblong in nature (see green and magenta arrows in Fig. 2B).
ical SEC-MALS setup allowing SAXS data collection
They were generally discrete in nature (not touching), but
from the individual protein peaks eluted from the size-
occasionally two oval domains would join at the base (see
cyan arrow in Fig. 2B). The arrangement of the domains exclusion chromatography column (see Materials and
relative to one another varied widely across the classes. Methods). SEC-MALS-SAXS measurements demon-
The size of the observed domains is consistent with the strated that EGFR COBRA is a well-folded and stable
size of individual immunoglobulin (Ig) domains, and their monomeric protein with a shape deviating from globular
round vs. oblong nature may correspond to top vs. side- (Supplementary Fig. S1, Table 1). We analyzed SAXS
views (Fig. 2C). Where two oval domains come together, data collected on the monomer of EGFR COBRA
the resulting shape is consistent with the projection of VH (Fig. 3A). The bell shape of the normalized Kratky plot
and VL pair forming an Fv (Fig. 2C, bottom). Diversity of indicates that EGFR COBRA is well folded (Fig. 3B).
composition and orientation of individual domains within The shift of the most prominent peak from the position
rows and clusters could come from significant conforma- of the most globular compact particle (the gray line at
√
tional variability of the protein or may represent different q.Rg = 3) suggests some disorder in EGFR COBRA or
views of the same particle. No more than five domains were the presence of a flexible unfolded region. The presence
visualized close together in a single particle. Two of those of the shoulder at q.Rg ∼ 6 indicates a multidomain
domains are likely engaged in Fv-like interactions but it arrangement (Fig. 3B). Furthermore, the asymmetrical
is impossible to identify which VH-VL pair is involved in peak in the pair distribution function P(r) plot suggests that
these interactions and which one is not. Two out of the EGFR COBRA has a hollow character with the Ig domains
seven Ig domains in EGFR COBRA were either disordered arranged in a circle rather than in a linear arrangement. The
or very flexible, or they may have been hidden from view. P(r) tail and its maximal dimension (Dmax) of 12.5 nm
Thus, we found that EGFR COBRA is a very flexible suggest that the molecule is elongated, likely featuring a
molecule, and some domains are likely changing their posi- protrusion or extension, resulting from a part or parts of
tion relative to each other by considerable distance without the structure not interacting with the core (Fig. 3C and
any preferred conformational state. Table 1).Antibody Therapeutics, 2022 7
In summary, the EGFR COBRA holds its shape well From this experiment we concluded that the MMP9
even though there is noticeable flexibility within the linker is presented in the structure of different COBRAs
molecule, most likely resulting from loose or dynamic very similarly. It is well exposed and positioned away from
interdomain interactions. the HSA and target binding sites. Similar extent of exposure
of the MMP9 linker observed in different COBRAs sup-
ports our conclusion from SAXS studies that all COBRAs
SEC-MALS-SAXS analysis of other COBRAs tested are folded similarly. Changing of sdAbs in the first
and second positions does not affect the overall fold of the
We proceeded to collect and analyze SAXS data obtained molecule.
on samples of other COBRAs, where EGFR targeting
sdAbs were replaced with ones targeting different tumor
proteins, to understand whether these substitutions affect Solution model of EGFR COBRA
the COBRA fold.
The MW determined by MALS showed that all COBRAs We attempted to build the three-dimensional model of
are monodisperse in the monomer state. The SAXS curves EGFR COBRA using the structural information we have
Downloaded from https://academic.oup.com/abt/article/5/1/1/6463575 by guest on 28 January 2022
(Fig. 3A) and their Rg values (Table 1) are similar to the collected by SAXS and negative stain EM.
EGFR COBRA data. P(r) functions for all COBRAs had Initially, SAXS data were used to calculate the ab initio
the same shape, indicating similarly shaped molecules in envelope of EGFR COBRA (see Materials and Methods)
solution (Fig. 3C). Analogously to EGFR COBRA, the (Fig. 4). The shape of the envelope was filled with the
other COBRAs also displayed some degree of flexibility atomistic models of EGFR COBRA individual domains
even though they were well folded (Fig. 3B). calculated based on the homology of these domains to
From SAXS data, we concluded that all COBRA Ig domains with known crystal structures (see Materials
molecules tested are shaped similarly and, most likely, are and Methods). The domains were then manually translated
folded in a similar way. The nature of the tumor targeting within the shape of the molecular envelope. Their positions
sdAbs in their first and second positions does not affect the were constrained by the order of the domains and the length
overall COBRA fold. of the flexible inter-domain linkers. By design, we expected
to find two pairs of VH and VL interacting with each other
in Fv-like orientation. However, we could not fit all the
domains into the SAXS envelope with four of them making
MMP9 linker exposure in EGFR COBRA and other
two pairs with Fv-like orientation without extending the
COBRAs
linkers beyond the physically permissible lengths. In the
To further address whether the fold of COBRAs remains negative stain EM images, we observed only one Fv-like
independent of the nature of sdAbs in the first and second pairing of two domains and only five domains were visible
position, we tested how well a common structural feature, simultaneously. Based on this observation, we released the
a linker cleavable by MMP9, connecting the second sdAb constrain of one Fv-like pair and built the model of EGFR
and VLi (Fig. 1), is exposed in a few different COBRAs. We COBRA shown in Figure 4 where only VH and VLi are
also tested whether binding of HSA or target protein affects engaged in Fv-like interactions. Unpaired VL and VHi
the cleavage of this linker in different COBRAs in solution. in this model appear to interact with other Ig domains
We prepared the MMP9 cleavage reaction mixtures with in uncommon orientations. In this model, the cleavable
COBRAs at the same protein molar concentrations and linker between the second sdAb and VLi resides on the
ratios, 2 μM of COBRA and 2 nM of MMP9. When the outer edge of the structure where it would be accessible by
cleavage reaction was performed in the presence of HSA or MMP9. The CDR loops on the sdAbs that are expected
target protein, they were added in excess to ensure satura- to engage in interactions with target proteins and HSA are
tion of the binding sites. The concentration of interacting also exposed and positioned away from the MMP9 cleav-
proteins exceeded their affinity constants, Kd , by three able linker (Fig. 4). The active VH and VL do not inter-
orders of magnitude. The reaction was carried out at room act with each other, congruent with the inactive prodrug
temperature and equal-sized aliquots were taken out at cer- conformation of the EGFR COBRA.
tain time points and analyzed for the relative concentration To test this model, we performed rigid-body modeling
of degradation products (Materials and Methods). Relative of the SAXS data using the program BILBOMD [22].
amounts of the cleaved COBRA (%) were then plotted on The initial model provided a moderate fit into the data
the kinetic curve and the initial velocities (rate of cleavage with χ 2 = 11.4 (Fig. 5A). However, when the multistate
of COBRA at the fixed initial protein concentration) were model generated by allowing the linkers extending from
compared. the N-terminal first sdAb and preceding the C-terminal
As shown in Figure 6 and Supplementary Figures S2–S5 aHSA sdAb to move was used in the refinement (Fig. 5B),
and Supplementary Table S1, the rates of MMP9 cleavage it resulted in a significant improvement of the SAXS fit.
of different COBRAs, measured as a slope of the initial part One- and two-state models gave an excellent match to the
of the kinetic curve, are very similar and are not affected SAXS data (χ 2 = 2.3 and 1.9), whereas a three-state model
by binding to HSA or target protein. Under conditions improved the SAXS-fit further to χ 2 = 1.1 (Fig. 5A).
of the experiment, where COBRAs were present at the To estimate a minimum number of conformational states
initial concentration of 2 μM and MMP9 at 2 nM, the in solution, we examined the Rg distribution [25] for the
initial velocity of cleavage reaction is about 1% of COBRA top 500 selected multistate models (Fig. 5A-inset). The Rg
per min. distribution of the two-state models has two broad peaks:8 Antibody Therapeutics, 2022
Figure 6. Initial velocity kinetics of MMP9 proteolytic cleavage of COBRAs. Accumulation of the cleaved product is plotted vs time.
one corresponding to closed conformations at Rg ∼36 Å were engaged in forming an Fv-like interface where these
and the other corresponding to open conformations at residues were buried.
Downloaded from https://academic.oup.com/abt/article/5/1/1/6463575 by guest on 28 January 2022
∼39 Å. For three-state models, the Rg distribution has These observations support the EGFR COBRA model
three significant peaks. The presence of multiple peaks in described above (Fig. 5B), with only one Fv-like pairing
Rg-distributions and the position of peaks validate the and implicate that this pairing is made between VH
flexibility of EGFR COBRA. and VLi.
The atomistic model shown in Figures 4 and 5B was not
the only model considered. An alternative model with an DISCUSSION
Fv formed by VL and VHi pairing could fit into the SAXS
envelope as well, although the domains would need to be In this study, we found that COBRAs are flexible and
rearranged relative to each other. Given the low-resolution dynamic molecules with a core made of a single Fv-like
structural information of SAXS and negative stain EM, we pairing between VH and VLi. The rest of the domains
were not able to distinguish between the models. However, are arranged around the core and move reasonably freely
all the considered models allowed only one Fv-like pairing relative to each other. Uncoupling of the VL and VHi
to fit into the SAXS molecular envelope. The model shown pair does not cause destabilization of the fold. Revealed by
in Figure 5B agreed excellently with the SAXS data after both SEC-MALS-SAXS and negative stain EM, COBRAs
accepting the reasonable assumption of the flexibility of the show very little, if any, signs of aggregation at the con-
N- and C-terminal linkers and has been evaluated further. centrations used in the experiments (up to 8 mg/ml, or
Thus, atomistic modeling of the SAXS data supports a 80 μM). Both techniques were also in agreement on the
COBRA fold with one Fv-like pairing. estimate of the longest dimension, measured at 12.6 nm by
SAXS and 12–15 nm by EM. At least in one orientation,
COBRA molecules appear to have an elongated shape while
maintaining a distorted globular overall shape.
Posttranslational modifications in EGFR COBRA
Judging by the shape of p(r) distribution function, all
As a protein molecule folds into its tertiary structure, COBRA molecules tested are shaped similarly suggesting
certain surfaces of its domains become inaccesible, and that switching of target-binding sdAbs has no impact on
others become exposed. The residues on the exposed, the overall fold of the molecule. Kinetic measurements and
solvent-accessible surfaces are more prone to posttransla- evaluation of exposure of the MMP9 linker led to the same
tional modifications (PTMs). We assessed PTMs in EGFR conclusion. In solution, MMP9 cleaves different COBRAs
COBRA using a mass spectrometry peptide map method. at the same rate and the cleavage is not affected by binding
EGFR COBRA was digested with α-chymotrypsin and of HSA or target proteins to the sdAbs. These observations
the mass of the resulting peptides was determined by mass support a model of VH-VLi Fv-like pairing forming a core
spectrometry and compared with the predicted mass of α- of the three-dimensional COBRA fold, while the other
chymotrypsin proteolytic fragments. We achieved > 95% domains have minimal interactions with this core and have
coverage of the polypeptide sequence of EGFR COBRA. positional flexibility. While in solution, the MMP9 cleavage
Among the PTMs identified, our attention was drawn to rates address the question of the COBRA fold, they may be
modifications of the residues in CDR3 loops of VL and influenced by other factors in more complex cellular and in
VHi domains that were not found in the CDR3 loops of vivo systems, such as by target density, presentation of the
VLi and VH domains (Supplementary Table S2). Given antigen epitopes, accessibility of the cell surface and others.
that residues of VL and VHi that acquired PTMs are Assessment of COBRA cleavage in these systems is beyond
present in VLi and VH, PTMs found in one of them, but the scope of this study.
not the another, could result from a different exposure or Even though the COBRA domains that are not involved
environment. If VH and VLi, or VL and VHi engaged in forming an Fv appear to have some positional flex-
in Fv-like interactions, the CDR3 would be involved in ibility, they may interact with each other in a weak or
forming the Fv-like interface (Supplemental Fig. S6) and dynamic way. We designed a series of domain deletions
be less solvent-accessible. Since the CDR3 loops residues (not described here) and found that only the construct
were modified in VHi and VL, we concluded that these two of EGFR COBRA without VHi domain can be purified
domains were not interacting in Fv-like fashion. On the and remains functional. However, it is not as stable as its
contrary, since the CDR3 loops residues were not modified parental construct, indicating possible involvement of all
in VH and VLi, we concluded that these two domains VH and VL domains in interactions stabilizing the overallAntibody Therapeutics, 2022 9
COBRA fold. From a literature analysis, it appears that FUNDING
although VH and VL domains typically interact with each The described study was funded by Maverick Therapeutics,
other in Fv-like fashion, they can make other types of Inc., a wholly owned subsidiary of Takeda Pharmaceutical
interactions, such as VH-VH or VL-VL pairings, engaging Company Limited. SAXS data collection at SIBYLS is
residues positioned on other surfaces rather than CDR3 funded through DOE BER Integrated Diffraction Analysis
loops. These alternative interactions were observed in the Technologies (IDAT) program and NIGMS grant [P30
crystal structures of diabodies with dimeric diabody assem- GM124169-01], ALS-ENABLE.
bly in the asymmetric unit [10, 11]. The protein concentra-
tion in crystallization experiments is usually high and most
likely those interactions were imposed by crystal packing CONFLICT OF INTEREST STATEMENT
forces. However, these crystal structures indicate that there None declared.
are complimentary surfaces on VHs and VLs that can
interact with each other. In antibodies of IgG format, VH
and VL loosely interact with CH1 and CL domains of DATA AVAILABILITY
Downloaded from https://academic.oup.com/abt/article/5/1/1/6463575 by guest on 28 January 2022
the Fab region in a ball-and-socket joint fashion [26]. The The COBRA molecules and sequences thereof are propri-
interactions defining the angle of the elbow in the Fab of etary and cannot be shared publicly.
different antibodies vary considerably, however, they are
present. Here, we hypothesize that in COBRA, unpaired
VL and VHi are likely stabilized by interactions with other REFERENCES
domains in the same polypeptide chain. Thus, the overall
1. Spiess, C, Zhai, Q, Carter, PJ. Alternative molecular formats and
fold of COBRA with VH-VLi FV-like pairing in the core therapeutic applications for bispecific antibodies. Mol Immunol
is stabilized by other interdomain interactions even though 2015; 67: 95–106.
they appear to be transient and dynamic by nature. 2. Baeuerle, PA, Reinhardt, C. Bispecific T-cell engaging antibodies for
cancer therapy. Cancer Res 2009; 69: 4941–4.
3. Walseng, E, Nelson, CG, Qi, J et al. Chemically programmed
bispecific antibodies in Diabody format. J Biol Chem 2016; 291:
CONCLUSIONS 19661–73.
4. Root, AR, Cao, W, Li, B et al. Development of PF-06671008, a
In summary, our studies of EGFR COBRA by SEC- highly potent anti-P-cadherin/anti-CD3 bispecific DART molecule
MALS-SAXS and negative stain EM allowed us to propose with extended half-life for the treatment of cancer. Antibodies 2016;
a three-dimensional model for this molecule where instead 5: 6.
of a conventional diabody with VH-VLi and VL-VHi 5. Reusch, U, Harrington, KH, Gudgeon, CJ et al. Characterization of
CD33/CD3 tetravalent bispecific tandem Diabodies (TandAbs) for
Fvs, we have only one Fv between VH and VLi. The the treatment of acute myeloid Leukemia. Clin Cancer Res 2016; 22:
model is consistent with analysis of the PTMs in EGFR 5829–38.
COBRA and assessment of the exposure of MMP9 6. Carmichael, JA, Power, BE, Garrett, TPJ et al. The crystal structure
cleavable linker. According to the model, the MMP9 of an anti-CEA scFv Diabody assembled from T84.66 scFvs in
linker is positioned away from HSA and tumor target VL-to-VH orientation: implications for Diabody flexibility. J Mol
Biol 2003; 326: 341–51.
binding sites and maintains a similar exposure in other 7. Chothia, C, Novotny, J, Bruccoleri, R et al. Domain association in
COBRAs where EGFR-binding sdAbs are substituted immunoglobulin molecules. The packing of variable domains. J Mol
by different tumor antigen-binding sdAbs. SAXS and Biol 1985; 186: 651–63.
MMP9 cleavage analyses of four COBRAs agree that 8. Abhinandan, KR, Martin, AC. Analysis and prediction of VH/VL
packing in antibodies. PEDS 2010; 23: 689–97.
our model for EGFR COBRA can withstand the sdAb 9. Herold, EM, John, C, Weber, B et al. Determinants of the assembly
switch. The model supports the fold of the COBRA into an and function of antibody variable domains. Sci Rep 2017; 7: 12276.
inactive conformation where binding to CD3 on T-cells is 10. Perisic, O, Webb, PA, Holliger, P et al. Crystal structure of a
prohibited. Activation would require cleavage of the MMP9 diabody, a bivalent antibody fragment. Structure 1994; 2:
linker followed by reassociation of fragments into dimers 1217–26.
11. Kim, JH, Song, DH, Youn, SJ et al. Crystal structure of mono- and
of active and inactive fragments. The active fragments bi-specific diabodies and reduction of their structural flexibility by
dimerizing via VH and VL interaction would create the introduction of disulfide bridges at the Fv interface. Sci Rep 2016; 6:
CD3 binding sites for cytotoxic T-cell engagement. 34515.
12. Panchal, A, Seto, P, Wall, R et al. COBRA™: a highly potent
conditionally active T cell engager engineered for the treatment of
solid tumors. MAbs 2020; 12: 1792130.
SUPPLEMENTARY DATA 13. Lander, GC, Stagg, SM, Voss, NR et al. Appion: an integrated,
database-driven pipeline to facilitate EM image processing. J Struct
Supplementary Data are available at ABT Online. Biol 2009; 166: 95–102.
14. Sorzano, COS, Marabini, R, Velázquez-Muriel, J et al. XMIPP: a
new generation of an open-source image processing package for
electron microscopy. J Struct Biol 2004; 148: 194–204.
ACKNOWLEDGEMENTS 15. Dyer, KN, Hammel, M, Rambo, RP et al. High-throughput SAXS
for the characterization of biomolecules in solution: a practical
We are thankful to Dr. Robert Fletterick, Professor approach. Methods Mol Biol 2014; 1091: 245–58.
Emeritus at University of California, San Francisco, 16. Hura, GL, Menon, AL, Hammel, M et al. Robust, high-throughput
for review of our manuscript and scientific discussion. solution structural analyses by small angle X-ray scattering (SAXS).
Nat Methods 2009; 6: 606–12.
We thank our Maverick Therapeutics, Inc., and Takeda 17. Franke, D, Svergun, DI. DAMMIF, a program for rapid ab-initio
Pharmaceuticals Company Limited former and present shape determination in small-angle scattering. J Appl Cryst 2009; 42:
colleagues for insightful discussions of this work. 342–6.10 Antibody Therapeutics, 2022
18. Volkov, VV, Svergun, DI. Uniqueness of ab initio shape determina- 23. Schneidman-Duhovny, D, Hammel, M, Tainer, JA et al. Accurate
tion in small-angle scattering. J Appl Cryst 2003; 36: 860–4. SAXS profile computation and its assessment by contrast variation
19. Wriggers, W, Milligan, RA, McCammon, JA. Situs: a package for experiments. Biophys J 2013; 105: 962–74.
docking crystal structures into low-resolution maps from electron 24. Schneidman-Duhovny, D, Hammel, M, Tainer, JA et al. FoXS,
microscopy. J Struct Biol 1999; 125: 185–95. FoXSDock and MultiFoXS: single-state and multi-state structural
20. Waterhouse, A, Bertoni, M, Bienert, S et al. SWISS-MODEL: modeling of proteins and their complexes based on SAXS profiles.
homology modelling of protein structures and complexes. Nucleic Nucleic Acids Res 2016; 44: 424–9.
Acids Res 2018; 46: 296–303. 25. Schneidman-Duhovny, D, Hammel, M. Modeling structure and
21. Sali, A, Blundell, TL. Comparative protein modelling by satisfaction dynamics of protein complexes with SAXS profiles. Methods Mol
of spatial restraints. J Mol Biol 1993; 234: 779–815. Biol 2018; 1764: 449–73.
22. Pelikan, M, Hura, GL, Hammel, M. Structure and flexibility within 26. Chiu, ML, Goulet, DR, Teplyakov, A et al. Antibody structure and
proteins as identified through small angle X-ray scattering. Gen function: the basis for engineering therapeutics antibodies.
Physiol Biophys 2009; 28: 174–89. Antibodies (Basel) 2019; 8: 55.
Downloaded from https://academic.oup.com/abt/article/5/1/1/6463575 by guest on 28 January 2022You can also read

















































