SEGUL: An ultrafast, memory-efficient alignment manipulation and summary tool for phylogenomics
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Posted on Authorea 4 May 2022 — The copyright holder is the author/funder. All rights reserved. No reuse without permission. — https://doi.org/10.22541/au.165167823.30911834/v1 — This a preprint and has not been peer reviewed. Data may be preliminary.
SEGUL: An ultrafast, memory-efficient alignment manipulation
and summary tool for phylogenomics
Heru Handika1 and Jacob Esselstyn2
1
Louisiana State University and A&M College
2
Louisiana State University
May 4, 2022
Abstract
With increasing use of genomic sequencing technology, phylogenetic studies routinely require manipulating and summarizing
thousands of alignments. Currently available software for these tasks uses considerable computing resources and are designed
for users with substantial knowledge of command line applications. We develop SEGUL, a compiled, single-executable tool
for fast alignment manipulation and summary tasks. SEGUL runs native on Windows, Linux, and macOS, includes native
support for Apple ARM Macs, and offers fast execution times and low memory footprints regardless of dataset size, operating
system, and CPU architecture (i.e., ARM or x86 64 CPUs). SEGUL includes a user-friendly command line interface, safety
features, and extensive documentation to aid beginners, while also providing advanced features. Keywords: segul, alignment
manipulation, concatenation, phylogenomics, phylogenetics, bioinformatics
Introduction
Alignment manipulation (e.g., filtering, splitting, extracting, and concatenating) and summary (e.g., num-
ber of parsimony informative sites, percent missing data, etc.) are common practices in phylogenetic tree
estimation workflows (see Oliveros et al. 2019; Chan et al. 2020; Esselstyn et al. 2021). With the advance
of genomic sequencing, alignments have grown exponentially in the number and length of sequences, and
manipulating these alignments with commonly used software often demands considerable computational re-
sources. The tasks are typically carried out using an application written in an interpreted programming
language, such as Python (e.g., Borowiec 2016; Faircloth 2016), R (e.g.,https://github.com/chutter/FrogCap-
Sequence-Capture), or Perl (e.g., Kück and Longo 2014). The computational efficiency of this approach is
limited by the requirement of an interpreter running alongside the application, type inference at runtime,
and garbage collection memory management, resulting in a high memory footprint. To optimize computing
efficiency, alignment manipulation tools such as AMAS eliminate file checks (e.g., file format and sequence
character checking) and rely on users to ensure that input files conform to file-type standards. Nevertheless,
these programs still have a high memory footprint (Borowiec 2016). For instance, to concatenate 4,060 ali-
gnments (560 Mb file size, 221 taxa, 2,464,926 sites), AMAS used 2.3 Gb of Random Access Memory (RAM)
space. Similarly, goalign uses a compiled programming language and eliminates dependencies required at
runtime, but does not solve the high memory footprint because of how memory management is handled.
For instance, concatenating the same alignments in goalign used 3 Gb of RAM. An approach using a high
performance programming language is required to obtain fast execution and efficient memory usage while
providing safer file parsing algorithms, and minimizing dependencies required at runtime.
A fast, memory efficient, reduced dependency application for phylogenetic studies not only enhances research
efficiency and repeatability, but also improves accessibility for evolutionary biologists with limited computing
resources while reducing the carbon footprint of bioinformatics. Developing such applications, however,
often requires using a fast, compiled programming language that allows fine control over how data are
1managed in computer memory. The two commonly used programming languages that have the feature, C
Posted on Authorea 4 May 2022 — The copyright holder is the author/funder. All rights reserved. No reuse without permission. — https://doi.org/10.22541/au.165167823.30911834/v1 — This a preprint and has not been peer reviewed. Data may be preliminary.
and C++ require programmers to ensure valid memory access, correct variable type to store data, and no data
races (i.e., multiple cores/threads modify data concurrently), which make them challenging to use (Perkel
2020). These code correctness issues are difficult to prevent and represent common problems in phylogenetic
software (Darriba et al. 2018). Due to the nature of using these programming languages, phylogenetic software
development using C/C++ is usually focused on the most demanding parts of phylogenetic workflows, such
as raw sequence read cleaning and adapter trimming (e.g. Fastp (Chen et al. 2018)), contig assembly (e.g.,
SPAdes (Bankevich et al. 2012)), sequence alignments (e.g, MAFFT (Katoh et al. 2002; Nakamura et al.
2018)), and phylogenetic tree estimation (e.g., RAxML-NG (Kozlov et al. 2019) and IQ-TREE (Nguyen et
al. 2015; Minh et al. 2020)). The recently emergent programming language, Rust, offers a safe alternative to
C/C++ (Köster 2016; Perkel 2020). It comes with an efficient development tool, guarantees valid memory
access, does not require garbage collection, and prevents data races for multithreading applications. As a
compiled programming language, Rust has few dependencies at runtime (relies on only the operating system
standard library) and can be distributed as a single executable file. Developing alignment and summary
statistics tools using Rust promises efficient performance, while eliminating dependency issues at runtime.
Furthermore, reducing dependencies minimizes conflict with other applications when used as part of analysis
pipelines and leads to improved research reproducibility.
We developed the SEGUL application for alignment manipulation and summary. Our application includes
an informative terminal output, a log file, comprehensive error checking, and a growing list of features for
alignment manipulation and summarization tasks. We designed SEGUL with beginners in mind, while still
providing advanced command features for more experienced users. As such, SEGUL is suitable both for
research and teaching. It carries the benefits of the Rust programming language, which guarantees only valid
memory access and multithreading performance without data races.
Features, Implementation, and Usages
SEGUL is a compiled, single executable, command-line application and requires zero depen-
dencies to run on macOS and Windows. On Linux, it relies on only the GNU C Library
(GLIBC,https://www.gnu.org/software/libc/ ) which comes pre-installed with Linux distributions. Users can
install the pre-compiled executable provided in the source code repository or compile the application from
the source code (see the Software Availability section below). The latter installation method expands SE-
GUL platform support to any platform supported by the Rust programming language (https://doc.rust-
lang.org/nightly/rustc/platform-support.html ). The compiler also fine-tunes the resulting executable for the
user’s computer.
SEGUL development focuses on improving efficiency when working with thousands of alignment files, enab-
ling analysis even on lower-end laptops. We achieve this goal by improving execution time, reducing RAM
usages, and simplifying command structure. Multiple file output will always be stored in a directory, so that
users don’t have to write custom code to organize files. SEGUL does not automatically overwrite existing
files but does provide an overwrite option for automated phylogenomic pipelines. SEGUL features a modern
terminal output with information on the application input, processing stages, and output (see example in
Figure S1). For record keeping, input and output information are logged to a file. Both the terminal output
and a log file improve repeatability while offering transparency during task execution so that mistakes on
input files are caught quickly. For example, for alignment concatenation, SEGUL will provide information
about the file counts, input format, datatype of input files, and information about taxon counts, alignment
counts, and alignment lengths for the output file. Some functions, such as sequence ID renaming, offer a
dry-run option to check if the application parses the input IDs correctly before processing input files.
SEGUL supports sequence input and output files in NEXUS, FASTA, and relaxed-PHYLIP formats (both
sequential and interleaved versions). For NEXUS and PHYLIP inputs that contain the taxon and site counts
in the header file, SEGUL compares the taxon counts and site counts of the parsed sequences with the
information in the header and will throw an error and abort processing if the information does not match.
By default, SEGUL checks that sequence characters in input files contain only IUPAC characters for DNA.
2Users are required to pass “–datatype aa” if the inputs are amino acid sequences. For most SEGUL functions,
Posted on Authorea 4 May 2022 — The copyright holder is the author/funder. All rights reserved. No reuse without permission. — https://doi.org/10.22541/au.165167823.30911834/v1 — This a preprint and has not been peer reviewed. Data may be preliminary.
users can improve computing efficiency by using “–datatype ignore” to skip checking IUPAC validity of
character states. This convenient feature is particularly useful when running SEGUL on large datasets using
computers with limited computing power. It is also a way to save computing time if users have previously
used the same dataset as SEGUL input. When concatenating alignments, SEGUL enforces that all sequences
in each alignment are the same length. Whenever possible and safe to use, the application takes advantage
of multi-core processors without the user needing to input the number of cores. Rather, SEGUL assesses
the available cores and uses the optimum number given the tasks. These automatic resource allocations are
determined by the Rust Rayon library (https://docs.rs/rayon/latest/rayon/ ). All SEGUL-critical and some
non-critical functions are tested using the unittest system provided by the Rust programming language. We
implement a continuous integration system using GitHub Action (https://github.com/features/actions) to
automatically validate code changes and ensure that failures in the designed tests are publicly displayed in
the source code repository.
SEGUL has a growing list of features for alignment manipulation and summary statistics (Table 1). Summary
statistics can be computed for an entire dataset, each alignment, and each taxon in an entire dataset. The
statistics are accurate even when none of the individual alignments contain all of the taxa represented in
the collection of alignments (e.g., Esselstyn et al. 2021). SEGUL can also extract sequences based on the
sequence IDs provided by the users as a terminal input, a list in a file, or regular expression. Users can filter
alignments based on taxon completeness (input in decimal percentage), alignment length (site counts), or
the number or percentage of parsimony informative sites (PIS). Often it is useful to know the taxa that are
present in an entire dataset, particularly when receiving alignment files from third party sources. This task
is particularly tedious to perform for genomic datasets with thousands of alignment files. The SEGUL ”id”
function can quickly provide a list of unique sequence IDs (taxa) in an entire dataset. For concatenating
alignments, SEGUL writes both the concatenated alignments and partition settings. The partition settings
are available in RAxML and NEXUS formats, including codon model support. The NEXUS partition can
be written as a separate file or embedded in NEXUS formatted sequences as a charset block.
Table 1. A full list of SEGUL features and command examples, as of version 0.16.3.
Features Command examples
Alignment concatenation segul concat –input –output
Alignment filtering segul filter –input [filtering-options] –output
Alignment splitting segul split –input –input-part –outputa range of taxa, site, and character counts (Table 2). We downloaded the datasets either directly from the
Posted on Authorea 4 May 2022 — The copyright holder is the author/funder. All rights reserved. No reuse without permission. — https://doi.org/10.22541/au.165167823.30911834/v1 — This a preprint and has not been peer reviewed. Data may be preliminary.
original sources or using BenchmarkAlignments scripts (https://github.com/roblanf/BenchmarkAlignments).
For alignments that were provided as concatenated files, we split the sequences into loci using the SEGUL
split function based on the partition settings provided by the authors in the source datasets. We ran the test
on four different platforms. Three platforms were desktop computers each using a different operating system
(Linux, Windows, and MacOS) and were equipped with high performance and relatively recent hardware.
For the Windows system, we ran the test on Windows Subsystem for Linux (WSL, Table S1). Both the
native Linux and the WSL systems used identical hardware running openSUSE Linux and the WSL host
operating system, Windows 11, in dual-boot mode. To investigate how the performance of SEGUL and
AMAS were impacted when using limited computing power, we tested each application on an eight-year old
Macbook Air laptop equipped with a two-core, four-thread processor and four gigabytes of Random Access
Memory (RAM) (Table S1). All tests were run on quiet computers with minimal applications running in
the background. On Windows, we set the terminal that ran the applications on high-priority execution.
To compare SEGUL performance to AMAS, we ran each analysis ten times per dataset and platform. In
addition, we also conducted a “warm-up” run for each application before each test to fill the computer cache.
This ensured that performance was not impacted by an empty cache. We also re-ran the test if we detected
outliers in the results. For the concatenated alignment test, AMAS by default does not check the alignment
length and it never checks that input sequences contain only IUPAC characters, whereas SEGUL checks both.
Therefore, we also tested AMAS using the “–check-align” option and SEGUL using “–datatype ignore”.
Testing alignment length is often useful in avoiding invalid results caused by unaligned sequences and/or file
parsing errors. For SEGUL, using “–datatype ignore” eliminates expensive computation for checking IUPAC
validity. For all tests, whenever possible, we ran AMAS using multicore settings by inputting all available
cores in the test platforms. We measured the execution time in seconds (secs), RAM usages in Megabytes
(Mb), and the percentage of CPU usages using GNU Time (https://www.gnu.org/software/time/ ). All tests
were run using SHELL scripts. We then cleaned and summarized the test results using dplyr v1.07 and
plotted the results using ggplot2 v3.3.5 on R version 4.1.1. All raw data, SHELL scripts, and R code used
for testing are available on GitHub (https://github.com/hhandika/segul-bench).
Table 2. Dataset sources and alignment statistics.
Datasets Datatype Taxon counts Locus counts Site counts Dataset url
Chan et al. DNA 50 13181 6,180,393 dx.doi.org/10.5061/dryad
(2020)
Esselstyn et al. DNA 102 4040 5,398,947 dx.doi.org/10.5281/zenod
(2021)
Jarvis et al. DNA 49 3679 9,251,694 http://gigadb.org/dataset
(2014)
Oliveros et al. DNA 221 4060 2,464,926 dx.doi.org/10.5061/dryad
(2019)
Shen et al. Amino acid 343 2408 1,162,805 dx.doi.org/10.6084/m9.fi
(2018)
Wu et al. Amino acid 90 5162 3,050,198 dx.doi.org/10.6084/m9.fi
(2018)
Test results
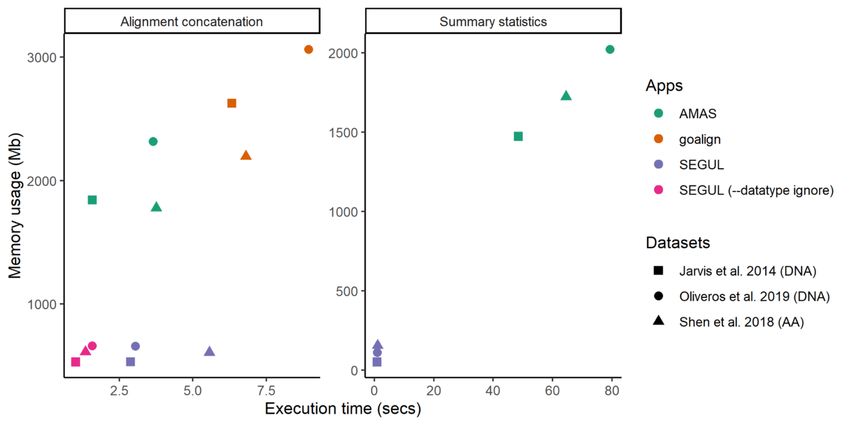
For alignment concatenation, on average across all tested platforms and datasets, SEGUL is 1.8 times faster
than AMAS, while using 0.33 of the RAM that AMAS used, both using the application default settings
(Table S2). SEGUL was faster than AMAS on all platforms, except Linux (Figure 1). On Linux, the default
AMAS concatenate function is 1.4 times faster than the default SEGUL concatenate function. However, we
used “–datatype ignore” in SEGUL to make the analyses more comparable, SEGUL is 2 times as fast as
4AMAS. On AMAS, if we used “–check-align” to make AMAS functionality comparable to default SEGUL
Posted on Authorea 4 May 2022 — The copyright holder is the author/funder. All rights reserved. No reuse without permission. — https://doi.org/10.22541/au.165167823.30911834/v1 — This a preprint and has not been peer reviewed. Data may be preliminary.
settings, SEGUL is 36 times faster than AMAS, and SEGUL with “–datatype ignore” option is 110 times
faster than AMAS (Figure S2). AMAS with “–check-align” option showed substantially slower execution
time when using datasets with many taxa, but the RAM usages remained the same. The RAM space usage
differences between SEGUL and AMAS were similar regardless of the settings and test platforms (Figure
1). Limited testing on Linux also showed that SEGUL with default settings, on average across all tested
datasets, was slightly faster (1.06 times) than goalign, while using 0.2 of the RAM that goalign used, whereas
the SEGUL with “–datatype ignore” was 2.1 times faster than goalign with a similar RAM difference as the
default SEGUL (Figure 1).
For the summary task, we were unable to run AMAS using multicore settings on WSL and Macbook Air
platforms. On WSL, AMAS never completed the task, whereas on Macbook Air, AMAS crashed the system
and forced a restart. Therefore, for these two platforms, we ran AMAS using a single core setting. On
average across all platforms and all datasets, SEGUL was 33 times faster than AMAS while using 0.03 of the
RAM space that AMAS used. SEGUL’s RAM usages were nearly equal across platforms, ranging between
68 to 90 Mb. SEGUL used the least amount of RAM on the Macbook Air platform (68 Mb average across
all tested datasets). AMAS’s RAM usage was substantially higher on WSL (7 Gb versus an average of 1.5
Gb in other platforms). This outlier could be caused by issues in WSL, the Python interpreter for WSL, or
both. We noticed similar behavior when we concatenated alignments on another Python program, Phyluce
(Figure S2).
Figure 1. SEGUL, AMAS, and goalign average execution time and RAM usage using three selected datasets
on Linux. SEGUL (–datatype ignore) is not available for summary statistics. Comparison using all datasets,
different settings, and different platforms are available in Figure S2 and Table S3.
Conclusions
SEGUL is an ultrafast, memory-efficient alignment tool to manipulate and generate summary statistics for
alignment files. It is consistently fast with low memory usages regardless of dataset, operating system, and
CPU architecture, while providing extra features, such as a log file, a more informative terminal output, and
more summary statistics. Its efficient use of computing resources and the inclusion of a log file offers greater
repeatability and accessibility than alternative applications.
Software Availability
SEGUL is open source and freely available under the Massachusetts Institute of Technology (MIT) license. It
is a cross-platform application and has been tested through automatic and manual testing on Windows (in-
cluding Windows Subsystem for Linux), Linux, and macOS (both on Intel and ARM CPUs). Pre-compiled
binaries and source code are available on GitHub athttps://github.com/hhandika/segul. SEGUL can also
5be installed using the Rust Package Manager, cargo, and is registered athttps://crates.io/crates/segul.
Posted on Authorea 4 May 2022 — The copyright holder is the author/funder. All rights reserved. No reuse without permission. — https://doi.org/10.22541/au.165167823.30911834/v1 — This a preprint and has not been peer reviewed. Data may be preliminary.
We provide extensive documentation on installing and using the application on the GitHub Wiki
athttps://github.com/hhandika/segul/wiki.
Acknowledgements
We thank Andre E. Moncrieff, Austin S. Chipps, Carl R. Hutter, Diego J. Elias, Giovani Hernández-Canchola,
Glaucia C. Del-Rio, Roberta C. Canton, Samantha L. Rutledge, Sarin Tiatragul, and Spenser J. Babb-
Biernacki for their feedback on the application and its documentation. Several SEGUL features are inspired
by Phyluce, AMAS, and FrogCap. SEGUL benefited greatly from general-purpose libraries, particularly
those provided by the Rust Programming Community.
Author Contributions
H.H. designed the application, wrote the code, and documentation. J. A. E. provide feedback on the appli-
cation design and wrote the documentation. Both authors wrote the manuscripts.
References
Bankevich, A. et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell
sequencing. Journal of Computational Biology , 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Borowiec, M. L. 2016. AMAS: a fast tool for alignment manipulation and computing of summary stati-
stics.PeerJ 4: 1660.https://doi.org/10.7717/peerj.1660
Chan, K. O., C. R. Hutter, P. L. Wood Jr, L. L. Grismer, and R. M. Brown. 2020. Target-capture phyloge-
nomics provide insights on gene and species tree discordances in Old World treefrogs (Anura: Rhacophori-
dae).Proceedings of the Royal Society B 287(1940). https://doi.org/10.1098/rspb.2020.2102
Chen, S., Y. Zhou, Y. Chen, and J. Gu. 2018. fastp: an ultra-fast all-in-one FASTQ preproces-
sor.Bioinformatics 34 (17): 884-890. https://doi.org/10.1093/bioinformatics/bty560
Darriba, D., T. Flouri, and A. Stamatakis. 2018. The state of software for evolutionary biology. Molecular
Biology and Evolution35:1037–1046. https://doi.org/10.1093/bioinformatics/btz305
Esselstyn, J. A., A. S. Achmadi, H. Handika, M. T. Swanson, T. C. Giarla, and K. C. Rowe. 2021. Fourteen
New, Endemic Species of Shrew (Genus Crocidura) from Sulawesi Reveal a Spectacular Island Radiation.
Bulletin of the American Museum of Natural History 454:1–108. https://doi.org/10.1206/0003-0090.454.1.1
Faircloth, B. C. 2016. PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinfor-
matics 32:786–788. https://doi.org/10.1093/bioinformatics/btv646
Jarvis, E. D. et al. 2014. Whole-genome analyses resolve early branches in the tree of life of modern birds.
Science 346:1320–1331. https://doi.org/10.1126/science.1253451
Katoh, K., K. Misawa, K.-I. Kuma, and T. Miyata. 2002. MAFFT: a novel method for rapid mul-
tiple sequence alignment based on fast Fourier transform. Nucleic Acids Research 30:3059–3066. htt-
ps://.doi.org/10.1093/nar/gkf436
Köster, J. 2016. Rust-Bio: a fast and safe bioinformatics library.Bioinformatics 32:444–446. htt-
ps://doi.org/10.1093/bioinformatics/btv573
Kozlov, A. M., D. Darriba, T. Flouri, B. Morel, and A. Stamatakis. 2019. RAxML-NG: a fast, scalable
and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35:4453–4455. htt-
ps://doi.org/10.1093/bioinformatics/btz305
Kück, P., and G. C. Longo. 2014. FASconCAT-G: extensive functions for multiple sequence alignment pre-
parations concerning phylogenetic studies. Frontiers in Zoology 11:81. https://doi.org/10.1186/s12983-014-
0081-x
6Minh, B. Q. et al. 2020. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the
Posted on Authorea 4 May 2022 — The copyright holder is the author/funder. All rights reserved. No reuse without permission. — https://doi.org/10.22541/au.165167823.30911834/v1 — This a preprint and has not been peer reviewed. Data may be preliminary.
Genomic Era. Molecular Biology and Evolution 37:1530–1534. https://doi.org/10.1093/molbev/msaa015
Nakamura, T., K. D. Yamada, K. Tomii, and K. Katoh. 2018. Parallelization of MAFFT for large-scale
multiple sequence alignments.Bioinformatics 34:2490–2492. https://doi.org/10.1093/bioinformatics/bty121
Nguyen, L.-T., H. A. Schmidt, A. von Haeseler, and B. Q. Minh. 2015. IQ-TREE: a fast and effective stocha-
stic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution32:268–274.
https://doi.org/10.1093/molbev/msu300
Oliveros, C. H. et al. 2019. Earth history and the passerine superradiation. Proceedings of the National
Academy of Sciences116:7916–7925. https://doi.org/10.1073/pnas.1813206116
Perkel, J. M. 2020. Why scientists are turning to Rust. Nature , 588:185–186.
https://doi.org/10.1038/d41586-020-03382-2
Shen, X.-X. et al. 2018. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell
175:1533–1545.e20. https://doi.org/10.1016/j.cell.2018.10.023
Wu, S., S. Edwards, and L. Liu. 2018. Genome-scale DNA sequence data and the evolutionary history of
placental mammals. Data in brief 18:1972–1975. https://doi.org/10.1016/j.dib.2018.04.094
7You can also read

















































