Neurodevelopmental disorders: 2021 update
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Free Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 1 of 23
Review
Neurodevelopmental disorders: 2021 update
Alfonsa Zamora‐Moratalla1, María Martínez de Lagrán1, Mara Dierssen1,2,3
1
Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, Dr. Aiguader 88,
Barcelona 08003, Spain
2
Universitat Pompeu Fabra (UPF), Barcelona, Spain
3
Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Spain
Corresponding author:
Mara Dierssen ∙ Systems Biology Program ∙ CRG‐Center for Genomic Regulation ∙ C/ Dr. Aiguader, 88 ∙ PRBB Building ∙ 08003
Barcelona ∙ Spain
mara.dierssen@crg.eu
Submitted: 23 February 2021 ∙ Accepted: 12 March 2021 ∙ Copyedited by: Cathryn Cadwell ∙ Published: 24 March 2021
Abstract
One of the current challenges in the field of neurodevelopmental disorders (NDDs) is still to determine their
underlying aetiology and risk factors. NDDs comprise a diverse group of disorders primarily related to neurode‐
velopmental dysfunction including autism spectrum disorder (ASD), developmental delay, intellectual disability
(ID), and attention‐deficit/hyperactivity disorder (ADHD) that may present with a certain degree of cognitive
dysfunction and high prevalence of neuropsychiatric outcomes. Last year, advances in human genomics have
begun to shed light on the genetic architecture of these disorders and large‐scale sequencing studies are starting
to reveal mechanisms that range from unique genomic DNA methylation patterns (i.e. “episignatures”) to highly
polygenic conditions. In addition, the contribution of de novo somatic mutations to neurodevelopmental diseases
is being recognized. However, progressing from genetic findings to underlying neuropathological mechanisms
has proved challenging, due to the increased resolution of the molecular and genetic assays. Advancement in
modelling tools is likely to improve our understanding of the origin of neurodevelopmental disorders and provide
insight into their developmental mechanisms. Also, combined in vivo editing of multiple genes and single‐cell
RNA‐sequencing (scRNA‐seq) are bringing us into a new era of understanding the molecular neuropathology of
NDDs.
Keywords: Autism spectrum disorder, ASD, Neurodegenerative disorders, Next generation sequencing, Microbiome, Preterm birth
Abbreviations bundle, ChIP‐seq ‐ Chromatin immunoprecipitation
sequencing, CNVs ‐ Copy‐number variants, CRISPR ‐
ADHD ‐ Attention‐deficit/hyperactivity disor‐ Clustered regularly interspaced short palindromic
der, ART ‐ Assisted reproductive technology, ASD ‐ repeats, DEGs ‐ Differentially expressed genes,
Autism spectrum disorder, ATAC‐seq ‐ Assay for DLPFC ‐ Dorsolateral prefrontal cortex, EEG ‐ Elec‐
transposase‐accessible chromatin using sequencing, troencephalographic, E/I ‐ Excitation and inhibition,
Cas9 ‐ CRISPR‐associated protein 9, CB ‐ Cingulum ENCODE ‐ Encyclopedia of DNA Elements,
Copyright: © 2021 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/),
which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are
indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.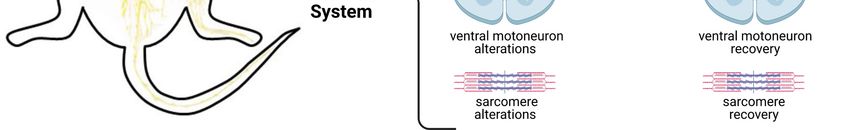
Free Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 2 of 23
EPT ‐ Extremely preterm, EWAS ‐ Epigenome‐wide proved challenging, due to the increased resolution
association studies, FLHS ‐ Floating‐Harbor syn‐ of the molecular and genetic assays. Advancement
drome, GEO ‐ Gene Expression Omnibus, GF ‐ Germ‐ in modelling tools is likely to improve our under‐
free, GO ‐ Gene Ontology, GW ‐ Gestational week, standing of the origin of neurodevelopmental disor‐
HD ‐ Huntington disease, hNPCs ‐ Human neural pro‐ ders and provide insight into their developmental
genitor cells, HTT ‐ Huntingtin, ID ‐ Intellectual disa‐ mechanisms. Also, combined in vivo editing of mul‐
bility, iPSCs ‐ Induced pluripotent stem cells, IQ ‐ In‐ tiple genes and single‐cell RNA‐sequencing (scRNA‐
telligence quotient, LGD ‐ Likely gene‐disruptive, lin‐ seq) are bringing us into a new era of understanding
cRNA ‐ Long intergenic noncoding RNA, mIPSCs ‐ the molecular neuropathology of NDDs.
Miniature inhibitory postsynaptic currents, mHTT ‐
Mutant HTT, MRI ‐ Magnetic resonance imaging, 1. Progress in the use of big data for
NDDs ‐ Neurodevelopmental disorders, NfL ‐ Neuro‐ the understanding the neuropathol‐
filament light protein, NLGs ‐ Neuroligins, NPCs ‐
Neural progenitor cells, nRT ‐ Nucleus reticularis ogy of neurodevelopmental disorders
thalami, NRXs ‐ Neurexins, PMDS ‐ Phelan‐McDer‐
Most NNDs have a major inherited component
mid syndrome, PSCs ‐ Pluripotent stem cells, preHD
which compromises correct brain development;
‐ Premanifest HD gene carriers, PRDD‐seq ‐ Parallel
however, the evaluation of the genetic causes of
RNA and DNA analysis after deep sequencing, PTMs
NDDs remains challenging because of genetic and
‐ Post‐translational modifications, REM ‐ Rapid eye
phenotypic heterogeneity. One well‐known exam‐
movement, RNA‐seq ‐ RNA‐sequencing, scRNA‐seq
ple is ASD, which has been associated with muta‐
‐ Single‐cell RNA‐seq, RSTS1 ‐ Rubinstein‐Taybi syn‐
tions in a wide range of genes, but relatively few
drome 1, sIPSCs ‐ Spontaneous inhibitory postsyn‐
genes or loci are identified in sufficient cases to
aptic currents, SPF ‐ Specific pathogen free, SWDs ‐
prove statistical significance at the genome‐wide
Spike‐wave discharges, VZ ‐ Ventricular zone, WES ‐
level. Such rare genetic developmental diseases are
Whole‐exome sequencing, WGS ‐ Whole‐genome
a challenge for diagnosis, as patients with the same
sequencing
genetic defect present with varying degrees of
symptoms and phenotypes (Haghshenas et al.,
Introduction 2020), that can be due to molecular interactions be‐
tween their associated genes, as in the case of Float‐
One of the current challenges in the field of ing‐Harbor syndrome (FLHS) and Rubinstein‐Taybi
neurodevelopmental disorders (NDDs) is still to de‐ syndrome 1 (RSTS1). In the last years, the fast pace
termine their underlying aetiology and risk factors. of development of next‐generation sequencing
NDDs comprise a diverse group of disorders primar‐ technologies, such as gene panels, whole‐exome se‐
ily related to neurodevelopmental dysfunction in‐ quencing (WES), and whole‐genome sequencing
cluding autism spectrum disorder (ASD), develop‐ (WGS) technologies have enhanced our ability to di‐
mental delay, intellectual disability (ID), and atten‐ agnose the NDDs. Of these, WES achieves a diagnos‐
tion‐deficit/hyperactivity disorder (ADHD) that may tic rate of 30–53% for NDDs and WGS improves the
present with a certain degree of cognitive dysfunc‐ diagnostic rates even more (42‐62%). The use of
tion and high prevalence of neuropsychiatric out‐ high‐throughput allows broader range of variant de‐
comes. Last year, advances in human genomics have tection, including noncoding and regulatory regions,
begun to shed light on the genetic architecture of and the discovery of novel disease‐associated genes,
these disorders and large‐scale sequencing studies has shed light on the genetic, genomic and epige‐
are starting to reveal mechanisms that range from nomic key players of NDDs and will also help in the
unique genomic DNA methylation patterns (i.e. “epi‐ diagnosis and stratification of the population with
signatures”) to highly polygenic conditions. In addi‐ those disorders. Changes in gene expression are also
tion, the contribution of de novo somatic mutations widely studied to characterize various human dis‐
to neurodevelopmental diseases is being recog‐ eases and successfully used to predict molecular and
nized. However, progressing from genetic findings cellular processes in complex neurodevelopmental
to underlying neuropathological mechanisms has diseases. High‐throughput expression profiling has
Free Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 3 of 23
become routine, and as a consequence, a vast Jensen et al. (Jensen et al., 2020), in an inter‐
amount of public RNA‐sequencing (RNA‐seq) da‐ esting study, added yet another level of complexity:
tasets has been generated and is available from the existence of comorbidities between NDDs. This
online repositories, such as Gene Expression Omni‐ is an important step, given the high degree of co‐oc‐
bus (GEO; Barrett et al., 2013). Up to the second currence of autism with ID that may blur the de novo
quarter of 2019, GEO hosted more than 112,000 variants and copy‐number variants (CNVs) identified
data series comprising more than 3,000,000 sam‐ in autistic individuals. The study analyzed 2290 indi‐
ples. This massive amount of biological data brings viduals from the Simons Simplex Collection for de
great opportunity for generating prominent biologi‐ novo likely gene‐disruptive (LGD) variants and CNVs,
cal hypotheses and is widely used to shed light into and determined their relevance regarding intelli‐
neuropathological mechanisms. However, these gence quotient (IQ) and Social Responsiveness Scale
large datasets were produced for diverse purposes, measures. By analyzing pathogenic de novo genetic
are sometimes difficult to interpret, and are not variants in individuals with autism who had either ID
friendly to large‐scale data integration. Therefore, or normal cognitive function, they determined
increasing sophistication in the statistical methods whether genes associated with autism also contrib‐
and well‐organized resources are required to enable ute towards ID comorbidity. Their study showed
efficient and extensive integrated analysis. that pathogenic de novo variants disrupting autism‐
associated genes contribute towards autism and ID
One interesting example is the work of Rahman comorbidity so that gene discoveries in autism are
et al., who performed a meta‐analysis using two biased towards genes that also contribute towards
publicly available RNA‐seq studies from ASD cere‐ comorbid ID (Jensen et al., 2020). In contrast, indi‐
bral cortex (Rahman et al., 2020). They performed viduals with high‐functioning autism are less proba‐
integrative RNA‐seq gene expression profiling in cor‐ ble to carry de novo LGD variants in candidate au‐
tex to identify transcriptional gene signatures al‐ tism genes and tend to present with disruption of
tered in 15 ASD subjects compared to 15 controls. genes with less functional relevance towards neuro‐
The study revealed core signatures of differentially development. These results highlight the relevance
expressed genes (DEGs) associated with ASD, includ‐ of dissecting phenotypic heterogeneity in family‐
ing already known markers of ASD and novel hub based sequencing studies of complex diseases. An‐
genes. The authors detected 235 unique DEGs, not other recent example is the identification of a non‐
identified by the individual studies, supporting the syndromic ASD subtype characterized by
increased statistical power of the meta‐analysis ap‐ dyslipidemia using massive multimodal data triangu‐
proach (Rau et al., 2014; Walker et al., 2008). Seven lation from WES, neurodevelopmental expression
of these DEGs (PAK1, DNAH17, DOCK8, DAPP1, PCD‐ patterns, electronic health records and healthcare
HAC2, ERBIN, and SLC7A7), were previously re‐ claims (Luo et al., 2020).
ported to be deferentially expressed in ASD. Gene
Ontology (GO) and pathways analysis was then used
as a tool for identifying molecular pathways en‐ 2. DNA episignatures
riched by the DEGs. In their meta‐analysis, Rahman
et al. showed altered osteoclast differentiation, TNF Genes associated with ASD are enriched for
signalling pathway, and complement and coagula‐ pathways affecting neuronal homeostasis and em‐
tion cascade pathways in ASD, revealing new previ‐ bryonic development. However, no single genetic
ously unidentified genes. Moreover, topological variant has been found that accounts for more than
analysis of protein–protein interaction of the ASD 1% of disease liability. This may be in part due to the
brain cortex revealed proteomics hub gene signa‐ fact that environmental factors are also known to
tures. However, although meta‐analysis is a power‐ contribute to ASD risk, especially during the prenatal
ful tool, it is also controversial due to the heteroge‐ period. Epigenetic mechanisms including DNA meth‐
neity in terms of platform (different microarray and ylation, histone post‐translational modifications
sequencing techniques), and sources (from periph‐ (PTMs), noncoding RNA, and chromatin architecture
eral blood to induced pluripotent stem cells [iPSCs] have been proposed to account for the sex bias,
or other tissues). gene‐environment interactions, and developmentalFree Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 4 of 23
origins of ASD aetiology (Ciernia & LaSalle, 2016). forebrain, including the assembly of the expanded
Epigenome‐wide association studies (EWAS) have human cerebral cortex, linked to the distinctively
identified ASD‐associated locus‐specific differential human features affected in NDDs, is a lengthy pro‐
methylation of genes involved in synaptic transmis‐ cess that involves diversification and expansion of
sion or microglia, and disease‐associated epigenetic neural progenitors, generation and positioning of
signatures (Nardone et al., 2014). EWAS in post‐ layer‐specific glutamatergic neurons, cellular migra‐
mortem brains have identified one of the most novel tion of GABAergic neurons, and formation and mat‐
and promising areas of research of NDDs, the iden‐ uration of glial cells. Disruption of these cellular
tification of DNA methylation called "episignatures". events by either genetic or environmental factors
Those are defined as the cumulative DNA methyla‐ can lead to neurodevelopmental disease, including
tion patterns occurring at multiple CpG dinucleo‐ ASD and ID. These complex cellular processes re‐
tides across the genome. quire highly synchronized regulatory activity under‐
lying these events, which, if perturbed, can cause
In the last years, an expanding number of ge‐ disease. The authors had previously used whole‐ge‐
netic syndromes have been shown to have unique nome bisulfite sequencing in placenta samples and
genomic DNA methylation patterns. The first clinical identified differential methylation of genes associ‐
genome‐wide DNA methylation assay, “EpiSign,” ated with ASD (Zhu et al., 2019). Now, they obtained
used genome‐wide DNA methylation analysis for the umbilical cord blood samples from ASD and typically
screening of 14 syndromes known to harbor ep‐ developing subjects from two high‐familial risk pro‐
isignatures. This first study showed that DNA meth‐ spective cohorts (i.e., cohorts following child’s early
ylation patterns are stable and specific to certain development of younger siblings of a child already
syndromes, and occur consistently across all of the diagnosed with ASD through) in order to identify an
individuals affected with the same syndrome (Aref‐ epigenomic signature of ASD at birth. Their findings
Eshghi et al., 2018). This has been confirmed re‐ suggest that epigenetic dysregulation in ASD may
cently by the identification of 34 disease‐specific ep‐ originate during early prenatal development in a
isignatures mapping onto 42 genetic syndromes, sex‐specific manner and converge on brain‐relevant
thus increasing the number of conditions that can genes to disrupt neurodevelopment. Although the
effectively be diagnosed through DNA methylation study has some limitations as the selection of high‐
testing (Aref‐Eshghi et al., 2020). The authors exam‐ familial risk cohorts and the limitation in statistical
ined emerging patterns of overlap, and similarities power, it opens a new framework for the prognosis
and hierarchical relationships across episignatures. and diagnosis of NDDs.
Aref‐Eshghi et al. implemented a uniform approach
for mapping DNA methylation signatures in numer‐
ous syndromes to enable unbiased comparisons. 3. Chromatin dynamics in neurodevel‐
This allowed identification of their key features as opment
they are related to genetic heterogeneity, dosage ef‐
fect, unaffected carrier status, and incomplete pen‐ As mentioned above, epigenetic gene regula‐
etrance. Through mass screening of a large cohort of tion plays a crucial role in controlling developmental
subjects with developmental delays and congenital transitions and cell differentiation, and is widely hy‐
anomalies, they demonstrate the utility of this tool pothesized to partly mediate risk for NDDs such as
in resolving ambiguous clinical cases and identifica‐ ASD or schizophrenia. Therefore, tracking epigenetic
tion of previously undiagnosed cases. changes in specific forebrain cell lineages over long
An interesting study has taken a slightly differ‐ time periods, has the potential to unravel the mo‐
ent approach. Mordaunt et al. using whole‐genome lecular programs that underlie cell specification in
bisulfite sequencing identified a distinct DNA meth‐ the human cerebral cortex and, by temporally map‐
ylation signature over regulatory regions and genes ping disease risk onto these changes, to identify cell
relevant to early fetal neurodevelopment in the cord types and periods of increased disease susceptibil‐
blood from newborns later diagnosed with ASD ity.
(Mordaunt et al., 2020). The development of theFree Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 5 of 23
In the last years, chromatin accessibility has a genome‐wide scale. However, one of the potential
emerged as an accurate proxy for the cellular regu‐ limitations of these methods is that you need to al‐
latory potential. Chromatin encodes epigenetic in‐ ready have an idea about what epigenetic mecha‐
formation in the form of post‐translational histone nisms are at play. Thus, the authors complemented
modifications and accessibility to DNA binding fac‐ this technique with assay for transposase‐accessible
tors (Allis & Jenuwein, 2016). Chromatin regulation chromatin using sequencing (ATAC–seq; Buenrostro
is affected by genetic alterations causative of NDDs et al., 2013), optimized for use on frozen tissues, to
(De Rubeis et al., 2014; Pinto et al., 2014; LaSalle, assay chromatin accessibility. ATAC‐seq identifies
2013), suggesting the existence of two classes of accessible DNA regions by probing open chromatin
NDDs. The first class is produced by mutations in with hyperactive mutant Tn5 transposase (Picelli et
chromatin regulators, as occurs in Rett syndrome al., 2014) that inserts sequencing adapters into open
(Schmidt et al., 2020). Importantly, those may target regions of the genome. The authors systematically
several convergent molecular axes, with genes ei‐ mapped chromatin state and accessibility across 72
ther belonging to the same class (e.g. lysine deme‐ distinct tissue‐stages of mouse development, and
thylases) or operating in the same molecular path‐ carried out integrative analyses incorporating addi‐
way (e.g. Polycomb‐mediated chromatin regula‐ tional epigenomic and transcriptomic data sets from
tion). The second class comprises those NDDs the same tissue‐stages. Importantly, their analysis
caused by environmentally‐induced epigenetic dys‐ allowed them to integrate chromatin state annota‐
function. Interestingly, accessible chromatin regions tions, infer the identities of dynamic enhancers and
exhibit a high heritability enrichment, indicating that key transcriptional regulators, and characterize the
sequence conservation can further refine functional relationship between chromatin state and accessi‐
risk genetic variants for disorders with a strong neu‐ bility during developmental gene regulation. As
rodevelopmental component, such as schizophrenia such, they could identify target genes and demon‐
(Bryois et al., 2018). strate tissue‐specific enrichments of variants associ‐
ated with disease in humans. Approximately 1–4%
Efforts to define the transcriptomic and epige‐ of the genome differed in chromatin state between
nomic landscapes of the developing human fore‐ tissues at the same stage, and 0.03–3% differed be‐
brain included multilevel analyses with characteriza‐ tween adjacent stages of the same tissue. This re‐
tion of spatiotemporal gene expression in the cortex source will help to map genetic risk for disease and
(Pollen et al., 2015; Nowakowski et al., 2018), the to shed light into gene‐regulatory dynamics at pre‐
molecular signature of cortical progenitors (Johnson viously inaccessible stages of human forebrain de‐
et al., 2015; Pollen et al., 2015), and epigenetics of velopment, including signatures of neuropsychiatric
early brain development (Amiri et al., 2018). Now, disorders. Data from this and all phases of ENCODE
Gorkin and partners of the Encyclopaedia of DNA El‐ are publicly available through the ENCODE portal
ements (ENCODE) project, have launched an atlas of (https://www.encodeproject.org).
chromatin development (Gorkin et al., 2020), a ge‐
nomic resource profiling epigenomics of mammalian Although encouraging, data on mouse devel‐
development. They initially used a diverse panel of opment do not completely mimic human forebrain
mouse tissues at 8 developmental stages from 10.5 development, which is, to a large extent, inaccessi‐
days after conception until birth, including transcrip‐ ble for cellular‐level study. The lack of availability of
tomes, methylomes and chromatin states to system‐ primary brain tissue samples and the limitations of
atically examine the state and accessibility of chro‐ conventional in vitro cellular models have precluded
matin in the developing mouse fetus. To map chro‐ a detailed mechanistic understanding of corticogen‐
matin states, the authors performed chromatin im‐ esis in disease states. Using long‐term three‐dimen‐
munoprecipitation with sequencing (ChIP–seq) for a sional (3D) directed differentiation of human plu‐
set of eight histone modifications that can distin‐ ripotent stem cells (PSCs) into dorsal and ventral
guish between functional elements and activity lev‐ forebrain domains as well as primary brain tissue
els. Methods like chromatin immunoprecipitation samples, a recently published work (Trevino et al.,
and reduced representation bisulfite sequencing al‐ 2020) found that organoids intrinsically undergo
low the investigation of epigenetic modifications on chromatin state transitions that are closely relatedFree Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 6 of 23
to human forebrain development. Trevino et al., the genomes of prokaryotic organisms such as bac‐
also applied ATAC‐seq in combination with RNA‐seq teria and archaea, which are used to detect and de‐
to map the epigenetic and gene expression signa‐ stroy DNA from similar bacteriophages during sub‐
tures of neuronal and glial cell lineages over 20 sequent infections. Cas9 (or "CRISPR‐associated pro‐
months in vitro. The authors identified epigenetic al‐ tein 9") is an enzyme that uses CRISPR as a guide to
terations putatively driven by specific transcription recognize and cleave specific strands of DNA. Cas9
factors and discovered a dynamic period of chroma‐ enzymes together with CRISPR sequences form the
tin remodeling during human cortical neurogenesis basis of the CRISPR‐Cas9 technology, used for gene
identifying key transcription factors that may coor‐ editing. CRISPR targeting in vivo, especially in mam‐
dinate over time to drive these changes. This ap‐ mals, can be difficult and time consuming when at‐
proach may bring new insights into gene‐regulatory tempting to determine the effects of more than a
dynamics at previously inaccessible stages of human single gene. However, such studies may be required
forebrain development, including signatures of neu‐ to identify pathological gene variants with effects in
ropsychiatric disorders. specific cells along a developmental trajectory. Pool
gene targeting followed by single‐cell RNA‐sequenc‐
ing of perturbed cells in the brain is a powerful
4. Sequencing perturbed cortex devel‐ methodology to reveal traits of individual cells in
opment heterogeneous populations such as those in brain
tissue (Dixit et al., 2016). Perturbations with single‐
The growing number of genetic disruptions cell sequencing readouts with increased throughput
identified in human genetic studies far exceeds the and enhanced resolution offer the possibility to ex‐
ability to study their functions, or perform rigorous plore the dynamics of transcription factors in devel‐
genotype‐phenotype correlations, which may vary opment and genes whose expressions are differen‐
substantially across different cell types and states. tial between cell states or across the different areas
Until now, genetic screens have systematically ana‐ of a tissue.
lyzed individual gene function in mammalian cells or Perturb‐seq was developed in 2016 (Dixit et al.,
in vivo in knockout models, analyzing each perturba‐ 2016), involving CRISPR/Cas9 to perform multi‐locus
tion separately. Also, screens have been performed gene perturbation with massive parallel scRNA‐seq.
in a pooled format, measuring, for example, cell au‐ As cellular behaviors typically depend on coordi‐
tonomous phenotypes, such as growth, drug re‐ nated expression of many genes and translated pro‐
sistance, or marker expression. Both screening teins, unbiased sequencing methods can extract ge‐
methods are time and labor intensive and do not al‐ nome‐wide profiles from single cells without prior
low the study of genetic interactions. Comprehen‐ knowledge. Jin et al. (2020) have now used Perturb‐
sive analysis of genetic interactions has been per‐ Seq to explore the effects of in vivo genetic disrup‐
formed in yeast between pairs of genes (Costanzo et tions of risk genes of ASD or NDDs across diverse
al., 2016). In mammals, only small sets of pre‐se‐ cells in the developing mouse cortex combined with
lected pairs have been assessed for cell viability single‐cell transcriptome sequencing. They evalu‐
(Bassik et al., 2013) or morphology (Laufer et al., ated 35 ASD de novo loss‐of‐function risk genes in
2013), but very few studies have examined higher multiple mouse embryos, using CRISPR‐Cas9 to in‐
order interactions or coupled those with a high con‐ troduce frameshift mutations in pools of these risk
tent scalable readout. genes. This allowed them to investigate how diverse
mutations affect cell types and states in the devel‐
The newest addition to the genomic arsenal is
oping organism. This method identified networks of
single‐cell clustered regularly interspaced short pal‐
gene expression in neuronal and glial cells that sug‐
indromic repeats (CRISPR) screening techniques, in‐
gest new functions in ASD‐related genes. Using
dependently termed Perturb‐Seq, CRISP‐seq, or
weighted gene correlation network analysis, they
CROP‐seq, that combine pooled CRISPR screening
identified 14 covarying gene modules representing
with scRNA‐seq to allow functional CRISPR screening
transcriptional programs in different types of corti‐
in single‐cells. CRISPR are DNA sequences found in
cal cells that affect common biological processesFree Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 7 of 23
across multiple cell types and/or represent cell question, researchers are using next‐generation sin‐
type–specific features. Perturbations in nine of gle cell functional genomic techniques, which enable
those ASD/NDD genes had significant effects across comprehensive and unbiased characterization of
cortical projection neurons, cortical inhibitory neu‐ the progenitor populations from the early fetal stage
rons, astrocytes, and oligodendrocytes. One exam‐ to neuronal maturation.
ple is Ankyrin, which interacts with ion channels in
excitatory neurons and stabilizes GABAergic synap‐ To elucidate the lineage relationships among
ses. Ank2 mutants show misregulation of intracellu‐ different cell types during brain development, last
lar calcium homeostasis and calcium channel ex‐ year Huang and colleagues (Huang et al., 2020) de‐
pression in excitatory neurons, and ectopic connec‐ veloped a novel method in which parallel RNA and
tivity, but now Perturb‐Seq data could identify addi‐ DNA analysis after deep sequencing (PRDD‐seq) al‐
tional roles of Ank2 in those interneurons co‐ex‐ lows simultaneous reconstruction of neuronal cell
pressing the Ndnf gene. Oligodendrocytes and as‐ type, cell lineage, and sequential neuronal for‐
trocytes were also affected by multiple risk gene mation in post‐mortem human cerebral cortex. This
perturbations. For example, Chd8 modulates oli‐ innovative approach revealed some conserved hu‐
godendrocyte differentiation and maturation, by di‐ man cell lineage patterns, including that inhibitory
rectly interacting with oligodendrocyte maturation and excitatory neurons diverge early in humans, and
genes. As highlighted in a comment on this work that excitatory neurons form following a similar “in‐
(Treutlein & Camp, 2020) in vivo Perturb‐Seq can re‐ side‐out” order as seen in animal models. Their work
veal neuronal and glial effects of sets of ASD/NDD shows that at least some human neural progenitor
risk genes associated with autism. cells (hNPCs) demonstrate restricted cell type out‐
put and that excitatory and inhibitory neurons are
generated from distinct progenitor regions, support‐
5. Study of lineage diversification in ing a model initially established in mice. The authors
the developing neocortex estimate that the number of progenitor cells that
generate the excitatory neurons within a cortical
As shown above, ASD susceptibility genes are column is ~10. By contrast, inhibitory neurons show
strongly interconnected and many act as genetic complex, subtype‐specific patterns of neurogenesis,
regulators of neurodevelopment of the cerebral cor‐ and not all of them are conserved relative to mouse.
tex. However, the core underlying neuropathologies
cannot be fully elucidated without understanding Using single‐cell RNA‐sequencing and in vivo
the cellular architecture of the human cortex, under‐ validation, Li et al. (2020) have now revealed previ‐
lying its susceptibility to disease. The cerebral cortex ously unrecognized neural stem and progenitor cell
is a complex structure formed by a wide repertoire diversity within the fetal mouse and human neocor‐
of neural cells shaping its unique configuration. In tex, including multiple types of radial glia and inter‐
only two areas of the adult mouse neocortex, single‐ mediate progenitors. This novel cellular diversity
cell transcriptomics has already identified at least 55 catalogue they describe is characterized by mixed
excitatory and 60 inhibitory neuron types (Ecker et transcriptional profiles and not so much by morpho‐
al., 2017; Tasic et al., 2016) and a highly diverse set logical classes, which may indicate unique functional
of excitatory and inhibitory neuron types that are or state‐dependent transcriptional profiles. In addi‐
mostly sparse, with excitatory types being less layer‐ tion, most of the previously unrevealed types of ba‐
restricted than expected in the middle temporal gy‐ sal progenitors have a human counterpart, indicat‐
rus of human cortex (Hodge et al., 2019). Such tre‐ ing again the inter‐species conservation of neurode‐
mendous cellular complexity of mature neurons is velopmental processes. Li et al. postulate that tran‐
especially intriguing, given that those come from a scriptional priming, a phenomenon whereby mRNA
limited number of progenitors. This apparent para‐ for proteins that will be expressed in progeny is pre‐
dox leads to one of the most crucial questions in sent (but not translated) in the parent cell, underlies
neurodevelopment: how the myriad of neuronal va‐ the diversification of a subset of ventricular radial
rieties located in the neocortex can arise from a re‐ glial cells in developing mouse and human brain (Li
duced number of progenitor types. To solve this et al., 2020). Transcriptional priming of radial glialFree Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 8 of 23
cells would generate specific types of basal progeni‐ on the rainbow mouse models. They developed a
tors and neurons, being thus a driver of precursor stable Cre‐inducible rainbow reporter hPSCline that
and lineage diversity. However, those studies still provides up to 18 unique membrane‐targeted fluo‐
elude the temporal scale in which progenitor popu‐ rescent barcodes. Because DNA recombination is
lations at the early foetal stage go through neuronal permanent the color barcode of a cell is inherited by
maturation. daughter cells during cell division, enabling determi‐
nation of the parental origin of cellular progeny and
Another recent study has also systematically tracking them longitudinally by live‐cell imaging. Us‐
profiled single‐cell transcriptome of the four cortical ing this technique, the authors have tracked neural
lobes and the pons. However, in this study they in‐ progenitor cells (NPCs) over a month of cortical neu‐
cluded the temporal dimension by analyzing tran‐ ron differentiation into 3D cultures. NPCs in the
scriptional landscapes from human embryo to mid monolayer culture had self‐organized into radially
gestation, covering more than 5 months of critical aligned cells forming a ventricular zone (VZ) in these
developmental stages (Fan et al., 2020). This strat‐ aggregates. The finding that monolayer cultures
egy allowed accurate temporal and spatial resolu‐ generate 3D cortical structures suggests that the dif‐
tion of developmental processes. As expected, the ferentiation protocol robustly recapitulates in vivo
authors observed marked distinctions between cer‐ signaling where the planar neural plate gives rise to
ebral cortex and the pons, in both molecular regula‐ the neural tube and its derivatives through morpho‐
tions and developmental patterns. The pons, an evo‐ genesis. In addition, longitudinal live imaging ena‐
lutionarily ancient structure, develops earlier show‐ bled determination of the timing of key neurogene‐
ing abundant interneurons at early embryonic sis events, such as VZ formation and neurite out‐
stages whereas in the cerebral cortex interneurons growth. These results strengthen the use of organ‐
start to appear beginning at the early mid‐fetal oids derived from human PSCs for the study of neu‐
stage, suggesting a delayed development of neurons rodevelopmental processes. Understanding cellular
in the cerebral cortex. Besides, molecular and elec‐ mechanisms that lead to dysregulated NPC expan‐
trophysiological results pointed out an asynchro‐ sion might help the understanding of neurodevelop‐
nous cell development in different regions of the mental disorders characterized by microcephaly or
cortex, with maturation occurring earlier in the ros‐ megacephaly.
tral regions than in the caudal ones, and regional dif‐
ferences on the lateral side of the developing cortex
appeared more conspicuous during the neuron mat‐ 6. Microbiome: the hidden player be‐
uration stage in the frontal lobe. The authors em‐ hind neurodevelopmental disorders
phasize the important role of long intergenic
noncoding RNA (lincRNA) in regional and cell type All the works presented above have revealed a
maintenance. These temporal differences in neu‐ remarkably complex molecular neuropathology in‐
ronal maturation could be essential for proper neu‐ volving a myriad of genetic architectures and regu‐
ral network construction and be beyond the estab‐ latory elements, including unexpected peripheral
lishment of certain neurodevelopmental disorders. players, such as the microbiota, in NDDs. The human
One limitation of this study, however, is that post‐ intestine harbors trillions of microbial cells which
mortem samples are collected at different gesta‐ form a symbiotic relationship with the host and play
tional ages only allow a raw estimation of the tem‐ a vital role in both health and disease (Huttenhower
poral evolution of cell lineages but not a longitudinal et al., 2012). The importance of microbiota to hu‐
tracing of the progenitors. man health is suggested by the observation that
A tool that tracks single‐cell lineages and their dysbiotic shifts in microbial communities have been
phenotypes longitudinally would reveal whether associated with a number of human diseases, in‐
heterogeneity extends beyond molecular identity. cluding obesity, inflammatory bowel disorders, or
This is exactly what El Nachef et al. (El‐Nachef et al., gastrointestinal cancer (Schulz et al., 2014; Flintoft,
2020) have now developed: a novel tool to track in 2012; Morgan et al., 2012; Qin et al., 2012). Recent
vitro dynamic behaviors at the single‐cell level based findings also suggest that gut dysbiosis can be in‐Free Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 9 of 23
volved in the development of a variety of neuropsy‐ based metagenome analysis have been reported for
chiatric disorders, such as ASD, depression and ASD gut microbiome (Laue et al, 2020; Wang et al,
schizophrenia (Grochowska et al., 2018). However, 2019), the high interindividual diversity, influenced
the high interindividual diversity due to genetics, by a wide range of factors such as genetics, age, diet,
age, diet, and health condition poses severe limita‐ and health conditions (Lloyd‐Price et al., 2016) im‐
tion to the study of ASD gut microbiome (Lloyd‐ pedes the correct identification of disease‐associ‐
Price, 2016), the results being highly dependent on ated microbiome features with stochastic false pos‐
the cohort studied. The composition of intestinal mi‐ itives or negatives and the findings are highly de‐
crobiome affects health from the prenatal period pendent on the samples collected (Surana & Kasper,
throughout adulthood, and should be thought of as 2017). To overcome this problem, a team of re‐
an organ system with important effects on child de‐ searchers in China proposed a new analytical strat‐
velopment (Ronan et al., 2021). In fact, gut microbi‐ egy, a quasi‐paired cohort analysis that successfully
ota plays a major role in the bidirectional communi‐ allowed them to identify a new gut microbe defi‐
cation between the gastrointestinal tract and the ciency in ASD (Zhang et al., 2020) children. In their
central nervous system and there is increasing evi‐ paper, Zhang et al. applied this novel strategy to a
dence of an important role of the gut microbiota in metagenomic study of the ASD microbiome. They
brain development and behavior (Hoban et al., collected stool samples from 39 children diagnosed
2016; Vuong et al., 2017; Warner, 2019). To date, with ASD, and 40 age‐ and sex‐matched neurotypical
most of our understanding of the interaction be‐ children of similar metabolic backgrounds, referring
tween the microbiota and the brain has come from to the profile of metabolic pathways inferred from
rodent models, in which the gut microbiota is linked the metagenomic data. This approach allowed them
to brain signaling mechanisms and affective pheno‐ to transform the original cohort into a paired cohort,
types, such as anxiety or depression‐like behavior. thus controlling for individual diversity and increas‐
ing statistical power. Each of the stool samples were
Animal studies have clearly demonstrated ef‐ subjected to metagenomic sequencing and the team
fects of the gut microbiota on brain gene expression focused most specifically on 18 microbial species
profiles and neurochemical metabolism impacting that have previously been linked to ASD. Using the
behavior and performance. Interestingly, transfer of quasi‐paired cohort analytical strategy, the authors
fecal gut microbiota from humans with depression identified significant deficiencies in detoxifying en‐
into rodents appears to induce animal behaviors zymes and pathways, strongly correlated with bi‐
that are hypothesized to indicate depression‐like omarkers of mitochondrial dysfunction in ASD chil‐
states (Kelly et al., 2016). Germ‐free (GF) animals dren. Then they applied diagnostic models based on
also show clear behavioral differences compared to these detoxifying enzymes, and could accurately dis‐
conventionally raised mice, or mice raised free of tinguish ASD individuals from controls of their co‐
specific disease‐causing organisms (specific patho‐ hort. In fact, the dysfunction score inferred from the
gen free; SPF) (Bäckhed et al., 2007), including social model was proportionally increased with the clinical
interaction disturbances similar to those of ASD; rating scores of ASD. These results were further
stress‐related and anxiety‐related behaviors; learn‐ proofed in an independent cohort of 65 ASD chil‐
ing and memory deficits; and impaired motor con‐ dren. These results suggest a gut microbiome impact
trol (Vuong et al., 2017). Some of the biochemical on detoxification in ASD, and a potential role of im‐
changes resulting from germ‐free status are irre‐ paired intestinal microbial detoxification in toxin ac‐
versible, even after colonization of the animals with cumulation and mitochondrial dysfunction, a core
normal gut microbiota later in life. Other abnormal‐ component of ASD pathogenesis (Castora, 2019).
ities, such as anxiety‐related behavior, can be ame‐ This is a very interesting finding as toxin exposure is
liorated after reconstitution of the gut microbiota one of the main etiological factors of ASD (Von Eh‐
(Clarke et al., 2013). renstein et al., 2019). The impaired detoxification
capability of the intestinal microbiome would reveal
Dysbiosis has been associated with pediatric a new mechanism explaining this increased vulnera‐
diseases, including autism, ADHD, asthma, and aller‐ bility to toxicant exposure of patients with ASD.
gies. However, although several studies of shotgun‐ Moreover, microbial detoxification capabilitiesFree Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 10 of 23
could thus be a new therapeutic target for patients adolescents, especially in low‐ and middle‐income
with ASD. countries, as diet is a potentially modifiable deter‐
minant of the functionality of gut microbiome
Emerging evidence suggests that the microbi‐ throughout life (Ghosh et al., 2020; Jebeile et al.,
ota’s influence on the central nervous system seems 2019; Liu et al., 2019; Sonnenburg & Sonnenburg,
likely to be established as traits early in life, and po‐ 2019). It is thus important not to neglect the later
tentially lead to later adverse mental health out‐ consequences of this crisis, forcing millions of fami‐
comes (Walker et al., 2008). In fact, the gut microbi‐ lies to rely on nutrient‐poor alternatives that may
ome development evolves throughout the lifespan not only lead to later obesity and diabetes, but also
from a composition that is simpler and more unsta‐ to a mental health pandemic (Pan et al., 2021).
ble during development to a highly diverse and sta‐
ble composition in the adult (Fouhy et al., 2012).
Even transient changes in gut microbial composi‐ 7. Neuroligin in neurodevelopmental
tion, induced by various intrinsic and extrinsic fac‐ disorders
tors, may drive enduring shifts in the microbiota
composition when the gut microbiota has not fully
matured or is generally unstable, such as during Alterations in synapse formation and function
early life. In fact, the vulnerability of microbiota are a hallmark of neurodevelopmental and neuro‐
composition during adolescence may explain why psychiatric disorders. Synapses are formed by a
this period shows increasing incidence of psychiatric complex process that is controlled by synaptic or‐
illnesses, including anxiety and mood disorders, psy‐ ganizer molecules, including trans‐synaptic adhe‐
chosis, eating disorders, personality disorders and sion molecules and secreted factors (Ferrer‐Ferrer &
substance abuse (Borre et al., 2014). The pathophys‐ Dityatev, 2018; Yuzaki, 2018). A key component of
iology of these disorders has been commonly inter‐ these organizational protein complexes are the syn‐
preted as arising from aberrations of maturational aptic adhesion proteins called neuroligins (NLGs), a
changes that normally occur in the adolescent brain. family of post‐synaptic cell‐adhesion proteins which
Now, a recent study (Lach et al., 2020) by Lach et al. act trans‐synaptically with neurexins (NRXs) another
has revealed that even transient microbiota deple‐ group of pre‐synaptic cell‐adhesion molecules. The
tion has long‐lasting effects on microbiota composi‐ NLGs and NRXs were one of the first pairs of synaptic
tion and leads to increased anxiety‐like behavior. adhesion molecules to be characterized, and their
The study was performed in mice exposed to antibi‐ mutation was quickly shown to be associated with
otic treatment for 3 weeks to achieve microbiota de‐ autism, and later with schizophrenia (Jamain et al.,
pletion during the adolescent period or in the adult‐ 2003; Südhof, 2008; Sun et al., 2011). Synaptic or‐
hood. This transient microbiota depletion had long‐ ganizational protein complexes play central roles
lasting effects on microbiota composition but only both in orchestrating synapse formation and in de‐
when antibiotics were administered in adolescent fining the functional properties of synaptic transmis‐
mice, in which it was associated with a significantly sion that together shape the flow of information
increased anxiety‐like behavior. To further under‐ through neuronal networks. However, although a
stand what pathways of communication between myriad of evidence supports the involvement of ge‐
the gut and the brain are responsible for such netic alterations in neuroligin genes, including point
changes at this time period, the authors analyzed mutations, missense mutations and internal dele‐
gene expression in the amygdala and again they tions in patients with ASD, their precise pathoge‐
showed that it was more severely affected in mice netic mechanism in NDDs still remains elusive. There
treated with antibiotics during adolescence. This are five members of the NLG family in humans and
highlights the vulnerability of the gut microbiota other primates: NLG1, 2, 3, 4X, and 4Y. Mutations in
during the adolescent period, influencing gene ex‐ NRX1, NLG3 and NLG4 genes are linked with ASD
pression and behavior even in adulthood. This is es‐ and other neurodevelopmental disorders (Parente
pecially important in the actual COVID‐19 crisis (Fore et al., 2017; Etherton et al., 2011; Sun et al., 2011;
et al., 2020; Headey et al., 2020), which poses severe Südhof, 2008; Dean & Dresbach, 2006; Lisé & El‐Hus‐
risks to the nutritional status of young children andFree Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 11 of 23
seini, 2006). Several NLGN2 variants have been iden‐ activity by electromyographic recordings. Interest‐
tified in individuals with schizophrenia, autism, and ingly, only NLG2 knockout mice exhibited abnormal
anxiety disorder (Parente et al., 2017). 5‐8Hz spontaneous spike‐wave discharges (SWDs),
an activity pattern that was correlated with behav‐
One longstanding hypothesis in the field is that ioral arrest episodes characteristic of absence sei‐
ASD arises from a disruption of the neuronal net‐ zures. Supporting their hypothesis that those arrest
work activity due to perturbation of the synaptic ex‐ episodes were reflecting absence, these were ame‐
citation and inhibition (E/I) balance. The NLGs family liorated by the anti‐absence seizure drug
regulates the balance between excitation and inhi‐ ethosuximide. This might result from the markedly
bition activity in the brain circuit by being able to reduced GABAergic and glycinergic inhibitory synap‐
modify the excitatory (NLGN4) / inhibitory (NLGN2) tic transmission, as assessed in the form of minia‐
balance. In particular, the NLG2 subtype is highly ex‐ ture or spontaneous inhibitory postsynaptic cur‐
pressed at inhibitory postsynaptic structures, and it rents (mIPSCs or sIPSCs) in NLG2 knockout mice. In
is required to establish appropriate inhibitory syn‐ fact, restoring GABAergic transmission by optoge‐
apse function during development, but also appears netic activation in a projection from the thalamic re‐
to play a role in synapse maintenance in adulthood ticular nucleus to the ventrobasal thalamus also par‐
(Liang et al., 2015; Troyano‐Rodriguez et al., 2019). tially normalized these phenotypes, indicating that
As such, NLG2 is a potent regulator of the E/I bal‐ they may result from loss of feedforward inhibition
ance: knockdown of NLG2 reduces GABA receptors in the circuit, the only region, together with the cen‐
and GABA‐mediated synaptic transmission, result‐ tral amygdala, reported to date to have near‐normal
ing in increased neuronal excitability (Liang et al., inhibitory synaptic transmission in the absence of
2015) while overexpression of NLG2 in transgenic NLG2 (Babaev et al., 2016). The same rescuing effect
mice leads to motor discoordination, social impair‐ was achieved by postsynaptic NLG2 expression in
ment and abnormal electroencephalographic (EEG) the thalamic neurons and optogenetic activation of
activities, characteristics shared by ASD and epilepsy the nucleus reticularis thalami (nRT) thalamic path‐
(Hines et al., 2008). This is interesting since high way was able to partially rescue GABAergic trans‐
prevalence of epileptiform activity emerges as a mission, SWDs, and behavior arrests in NLG2 knock‐
common pathophysiological hallmark of autism (Su‐ out mice.
bota et al., 2017; Horváth et al., 2016; Plioplys et al.,
2007). NLG2 might be thus a common player ex‐ Previous results have indicated that the imbal‐
plaining the comorbidity, as ASD and epilepsy fre‐ ance in the excitation/inhibition tone in the
quently coexist in the same individual, suggesting a thalamocortical circuitry is linked with the SWDs, ab‐
common neurodevelopmental basis for these disor‐ sence seizures and other forms of epileptic behav‐
ders. However, even though genome‐wide associa‐ iors (Fogerson & Huguenard, 2016; Maheshwari &
tion studies recently allowed for the identification of Noebels, 2014; Paz et al., 2011). The results of Cao
a substantial number of genes involved in ASD and and colleagues now provide strong evidence that
epilepsy, there was no direct evidence to support NLGN2‐mediated GABAergic transmission is a pow‐
the specific involvement of NLG2 in epileptic sei‐ erful regulator of thalamocortical network activities
zures. related to absence seizures and may provide a mo‐
lecular mechanism underlying high rates of comor‐
Now, in a recent paper, Cao et al. (Cao et al., bidity of ASDs with increased epileptic seizures,
2020) demonstrate that loss of NLG2 produces sei‐ providing a link between ASD phenotype and ab‐
zures and behavioral deficits that are associated sence seizures through a NLG2‐mediated inhibition
with GABAergic transmission in thalamic neurons. at the nRT‐thalamic synapse.
They systematically performed cortical EEG record‐
ings in freely moving NLG1, NLG2, and NLG3 knock‐
out mice and their wild‐type littermates in awake,
rapid eye movement (REM) sleep, and non‐REM
sleep states, simultaneously monitoring their motorFree Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 12 of 23
8. Are neurodegeneration diseases data of children and adolescent carriers of mHTT
showed that developmental trajectories in the stria‐
programmed from development? tum and globus pallidus were markedly different be‐
Since some years ago, accumulating evidence is tween gene‐expanded and gene‐non‐expanded in‐
revealing similarities and frequent complex interac‐ dividuals (Van Der Plas et al., 2019), a pattern that
tions between neurodevelopmental and neuro‐ was exaggerated with CAG expansion length > 50.
degenerative disorders. Neurodevelopmental con‐
ditions can manifest as lifelong weaknesses in cogni‐ The striking difference in developmental pat‐
tive functions and a number of works suggest that terns suggests that pathogenesis of HD begins with
certain neurodevelopmental disorders may entail abnormal brain development, where children who
greater risk for specific neurodegenerative disor‐ carry the gene expansion exhibit different trajecto‐
ders. An interesting example is Huntington disease ries of brain growth than those who did not inherit
(HD) a neurodegenerative disease characterized by the expansion. Now, Scahill et al. (2020) have
dyskinesia, progressive involuntary movements, be‐ showed that clinical biomarkers of HD neurodegen‐
havioral and psychiatric disturbances, and demen‐ eration such as cerebrospinal fluid neurofilament
tia. These symptoms are accompanied by wide‐ light protein (NfL) are already significantly elevated
spread neurodegeneration that likely starts in the as early as 24 years from predicted onset of clinical
striatum (Nopoulos, 2016) but also involves fronto‐ symptoms in a cohort of premanifest HD gene carri‐
striatal circuitry malfunction. It is prevalent in adult ers (preHD) young adults, in which motor, cognitive,
individuals, with a mean age at onset of ~45 years and psychiatric function are preserved,. A curious
(Langbehn et al., 2010) and initiates with motor dis‐ finding of Tereshchenko et al. (Tereshchenko et al.
abilities, such as involuntary movements. 2020) also indicates that measures of growth are ab‐
normal in child and adolescent carriers of mHTT dec‐
HD is caused by mutation of the huntingtin ades before HD onset. Specifically, gene expanded
(HTT) gene, as a result of CAG trinucleotide repeat males were taller than gene non‐expanded males in
expansion with repeat length ranging from 10 to 35 adolescents of similar weight.
in the normal population. Repeat lengths between
36 and 39 cause HD at reduced penetrance, and Furthermore, Barnat et al. (Barnat et al. 2020)
when expanded to 40 or more repeats, mutant HTT and DiFiglia (DiFiglia 2020), have gone even earlier,
(mHTT) causes HD at full penetrance. Despite symp‐ and studied human brain cortex of HD mutation car‐
toms of HD manifesting in adulthood, the aberrant rier fetuses and healthy controls at gestational week
HTT protein is present much earlier in persons car‐ 13 (GW13) and of a HD knock‐in mouse model at
rying the disease‐causing mutation. In fact, HTT is in‐ embryonic day 13.5 (E13.5), which correspond to
volved in brain development, axonal transport, syn‐ GW13. In this phase of development, progenitor
aptic function and cell survival and mHTT impairs cells in the VZ are extending processes towards the
NPC division, neuronal migration and maturation apical and basal surfaces of the neuroepithelial wall.
(Saudou & Humbert, 2016). Mutation of HTT in early The authors found, both in human fetuses and in
life is enough to induce HD features in adult mice mouse embryos, abnormalities in the developing
and shows that there is a developmental component cortex, including mislocalization of mHTT and junc‐
in the disease (Arteaga‐Bracho et al., 2016; Molero tional complex proteins, defects in NPC polarity and
et al., 2016) and HD mice present in fact thinner cor‐ differentiation, abnormal ciliogenesis, and changes
tices (Godin et al., 2010). There are a number of lines in mitosis and cell cycle progression. In normal de‐
of evidence that show cognitive function starts to velopment, NPCs in the VZ reach out to both the ap‐
decline and functional deficits appear around 15 ical and basal surfaces of the neuroepithelial wall,
years before clinical onset (Lee et al., 2012; Paulsen and their cellular nuclei shuttle back and forth as the
et al., 2008; Tabrizi et al., 2020). An interesting ap‐ cell cycle progresses. With the aberrant mHTT, epi‐
proach was taken by the Kids‐HD study, which in‐ thelial junctions are disrupted, epithelial polarity is
cludes a unique cohort of children and adolescents disturbed, and the cell cycle favors premature neu‐
who are at risk for adult‐onset HD (Lee et al., 2017). ronal differentiation. These data are in good agree‐
A recent study on this cohort using neuroimagingFree Neuropathology 2:6 (2021) Zamora‐Moratalla et al
doi: https://doi.org/10.17879/freeneuropathology‐2021‐3268 page 13 of 23
ment with previous experimental data that HTT reg‐ motor unit development using a combination of pa‐
ulates cellular adhesion, polarity, and epithelial or‐ tient‐derived human iPSCs, Shank3Δ11 (−/−) mice,
ganization (Godin et al., 2010). In summary, altera‐ and PMDS muscle biopsies from patients. The au‐
tions in neurodegenerative genes or proteins during thors show that hypotonia in Shank3 deficiency
developmental stages might program neurodegen‐ might be caused by dysfunction in all elements of
eration disorders in adulthood. However, given their the voluntary motor system: motoneurons, neuro‐
neurodevelopmental effects, it is unknown why muscular junctions and striated muscle fibers. These
these disorders do not emerge until adult stages. findings provide a new perspective on the function
of Shank3, beyond the central nervous system, and
9. Shank‐3: the missing link suggests a future treatment strategy for Shank3‐as‐
sociated ASD. Furthermore, recent evidence shows
The wide variety of NDDs that present with mo‐ that somatosensory neurons in Shank3‐mutated
tor symptoms is well established. In recent years, mice are dysfunctional and contribute to tactile phe‐
there has been an increased interest in the motor notypes (Orefice et al., 2019). Therefore, impaired
deficits of ASD in order to find a target to diagnose proprioception and altered somatosensory feed‐
the disease as early as possible in young children. back to the central nervous system of patients with
However, the landscape of motor deficits is quite PMDS and ASD may worsen symptoms and induce a
broad and may involve brain, peripheral nervous delay in motor development.
system or neuromuscular junction. ASD patients Abnormal functional communication between
may present with motor‐related abnormalities such brain areas has recently arisen as a feasible bi‐
as delayed motor development (Ozonoff et al., omarker for ASDs (Emerson et al., 2017; Lewis et al.,
2008), impairment of gross and fine motor functions 2013). Interestingly, Zhou et al. (2019) showed evi‐
(Provost et al, 2007) and deficits in basic motor con‐ dence of atypical behavior and altered brain circuits
trol (Jansiewicz et al., 2006). Some patients may in Shank3‐mutant macaques. The authors used
have trouble with coordinating movements be‐ CRISPR‐Cas9‐mediated gene‐editing technology to
tween the left and right side of the body, low muscle genetically engineer non‐human primate models
tone or balance. One possible candidate to explain (Macacafascicularis and their offspring), which
part of these symptoms is Shank3. Haploinsuffi‐ might better approximate the behavioral and neural
ciency of the Shank3 gene represents one of the phenotypes of ASDs than do rodent models. Mag‐
most common single‐gene mutations in ASD and has netic Resonance Imaging (MRI) in Shank3 mutants
been associated with neural circuit alterations in revealed altered local and global connectivity pat‐
several brain areas (Holder and Quach, 2016; Le‐ terns among brain areas that were indicative of cir‐
blond et al., 2014; Betancur et al., 2013). Shank3 is a cuit abnormalities. This was paralleled by social and
structural protein accumulated in postsynaptic den‐ learning impairments, sleep disturbances, repetitive
sities, which contributes to excitatory synapse func‐ behaviours and motor deficits. The works of Lutz et
tion and plays various roles in development (Wang al. (2020) and Zhou et al. (2019), together, make it
et al., 2014). Moreover, the Shank3 gene is the main clear that Shank3 protein is affecting the physiologi‐
contributing factor in Phelan‐McDermid syndrome cal functioning of the motor system from the cere‐
(PMDS) that is generally characterized by neonatal bral cortex to the nerve endings of the body's ex‐
skeletal muscle hypotonia despite normal growth, tremities.
severely delayed speech, moderate to profound de‐
velopmental delay, and mild dysmorphic features In addition to these findings, using multipara‐
(Sarasua et al., 2014; Kolevzon et al., 2014; Soorya metric MRI analysis by diffusion tensor imaging and
et al., 2013). Hypotonia is critical for diagnosis be‐ volumetry, it has recently been shown that the lack
cause it is one of the earliest clinical hallmarks of of Shank3 induces mainly changes in white matter
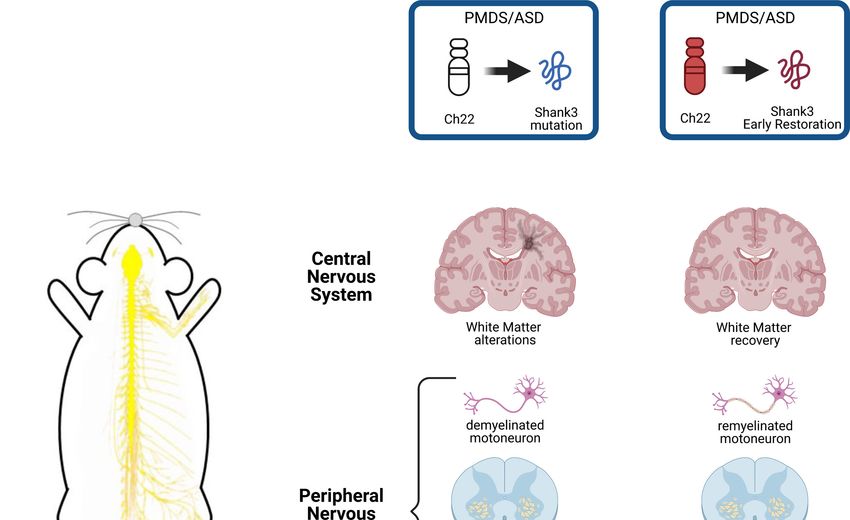
PMDS. Indeed Lutz et al. (Lutz et al., 2020) now re‐ (Jesse et al., 2019). In Shank3‐deficient patients with
ported the first experimental evidence that directly PMDS the authors found a significant pattern of
links the mutation of Shank3 with peripheral muscle white matter alterations in the long association fiber
atrophy. They investigated the role of Shank3 on tracts. White matter alterations were also observedYou can also read

















































