N-Acetylglucosamine drives myelination by triggering oligodendrocyte precursor cell differentiation
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
JBC Papers in Press. Published on September 25, 2020 as Manuscript RA120.015595
The latest version is at https://www.jbc.org/cgi/doi/10.1074/jbc.RA120.015595
N-Acetylglucosamine drives myelination by triggering
oligodendrocyte precursor cell differentiation
Michael Sy1, Alexander U. Brandt1,2,3, Sung-Uk Lee1, Barbara L. Newton1, Judy Pawling4,
Autreen Golzar1, Anas A. Rahman6, Zhaoxia Yu7, Graham Cooper2,3, Michael Scheel3,
Friedemann Paul2,3,8, James W. Dennis4,6, Michael Demetriou1,5$
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
1
Department of Neurology, University of California Irvine, 208 Sprague Hall, Mail Code 4032,
Irvine, CA 92697, USA.
2
Experimental and Clinical Research Center, Charité – Universitätsmedizin Berlin and Max-
Delbrueck-Center for Molecular Medicine, Lindenberger Weg 80, 13125 Berlin, Germany.
3
NeuroCure Clinical Research Center, Charité – Universitätsmedizin Berlin, corporate member
of Freie Universität Berlin, Humboldt-Universität zu Berlin and Berlin Institute of Health,
Charitéplatz 1, 10117 Berlin, Germany.
4
Samuel Lunenfeld Research Institute, Mount Sinai Hospital, 600 University Ave, Toronto, ON
M5G 1X5, Canada.
5
Department of Microbiology and Molecular Genetics, University of California Irvine.
6
Department of Molecular Genetics, University of Toronto, ON M5S 1A8, Canada.
7
Department of Statistics, Donald Bren School of Information and Computer Sciences
University of California Irvine, Bren Hall 2019, Irvine, CA 92697, USA
8
Department of Neurology, Charité – Universitätsmedizin Berlin, Charitéplatz 1, 10117 Berlin,
Germany.
$
Correspondence to: Michael Demetriou, Department of Neurology, University of California
Irvine, 208 Sprague Hall, Irvine, CA 92697, USA; Phone: (949) 824-9775, Email:
mdemetriou@uci.edu.
Running title: N-acetylglucosamine and myelination
Keywords: N-glycan branching, N-acetylglucosamine, oligodendrocytes, myelination, myelin
repair, multiple sclerosis.
1
Abstract oligodendrocytes is often incomplete despite
the presence of abundant oligodendrocyte
Myelination plays an important role in precursor cells (OPC) throughout the brain
cognitive development and in demyelinating (6-10). The molecular mechanisms that
diseases like multiple sclerosis (MS), where block re-myelination in MS are
failure of re-myelination promotes incompletely understood and there is a lack
permanent neuro-axonal damage. of therapies to promote myelin repair.
Modification of cell surface receptors with Failure to adequately re-myelinate is
branched N-glycans coordinates cell growth influenced by the microenvironment of the
and differentiation by controlling MS lesion, where reactive astrocytes,
glycoprotein clustering, signaling and microglia, and macrophages produce various
endocytosis. N-acetylglucosamine (GlcNAc) inhibitory factors leading to disruption in
is a rate-limiting metabolite for N-glycan OPC differentiation, oligodendrocyte
branching. Here we report that GlcNAc and migration, process outgrowth, and
N-glycan branching trigger attachment to axons (11). Multiple studies
oligodendrogenesis from precursor cells by have identified molecules that limit OPC
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
inhibiting PDGF receptor-α cell endocytosis. differentiation into myelin producing cells
Supplying oral GlcNAc to lactating mice including LINGO-1 (12), various
drives primary myelination in newborn pups extracellular matrix proteins (13,14), and
via secretion in breast milk, while myelin debris (15). Thus, increasing OPC
genetically blocking N-glycan branching differentiation has become an important
markedly inhibits primary myelination. In strategy for promoting remyelination in MS
adult mice with toxin (cuprizone) induced and other demyelinating diseases (16).
demyelination, oral GlcNAc prevents neuro-
axonal damage by driving myelin repair. In Cell surface and secreted proteins are co-
MS patients, endogenous serum GlcNAc and post-translationally modified on Asn(N)
levels inversely correlated with imaging by the addition of carbohydrates (N-glycans)
measures of demyelination and in the endoplasmic reticulum and
microstructural damage. Our data identifies subsequently remodeled in the Golgi. The
N-glycan branching and GlcNAc as critical degree of N-acetylglucosamine (GlcNAc)
regulators of primary myelination and branching in N-glycans promotes binding to
myelin repair and suggests oral GlcNAc galectins, a family of sugar-binding proteins
may be neuro-protective in demyelinating (Fig. S1A). Poly-valent galectin –
diseases like MS glycoprotein interactions at the cell surface
form a macromolecular lattice that
Introduction simultaneously controls the movement,
clustering and/or endocytosis of multiple
Myelination of axons by oligodendrocytes in receptors and transporters to control
the central nervous system plays an critical signaling, cell growth, differentiation and
role in normal cognitive development and death (17-24). For example, N-glycan
function as well as in demyelinating disease branching controls epithelial cell growth by
such as multiple sclerosis (MS) (1,2). In regulating receptor tyrosine kinases
addition to speeding conduction of the endocytosis (17-20), promotes glucose
action potential, myelination supports axon uptake in mesenchymal and pancreatic b
health and survival (3-5). In MS, re- cells by inhibiting glucose transporter
myelination of demyelinated axons by endocytosis (19,25) and reduces T cells, B
2
cell and neutrophil pro-inflammatory oligodendrocyte progenitor/precursor cells
responses by co-regulating the clustering (OPC)(42). In epithelial cells, N-glycan
and/or endocytosis of multiple glycoproteins branching deficiency reduces PDGFRα
(17,23,26-29). These mechanisms in turn surface expression by enhancing loss via
impact cancer, type II diabetes and endocytosis, leading to reduced signaling
autoimmunity (20). For example, reductions (18). Thus, here we examine the hypothesis
in N-glycan branching are associated with that GlcNAc may provide an oral
MS and promote both inflammatory therapeutic to raise N-glycan branching in
demyelination and neurodegeneration in OPC’s, promote myelination and reduce the
mice, the latter by an unknown mechanism potential for neurodegeneration by initiating
(17,30-36). oligodendrocyte differentiation via enhanced
PDGFRα surface expression and signaling
Given the diverse and pleiotropic effects of in OPC’s.
N-glycan branching, identifying and
manipulating regulatory mechanisms may Results
provide new insights into disease
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
pathogenesis and opportunities for GlcNAc and N-glycan branching trigger
therapeutic intervention. In this regard, oligodendrogenesis by inhibiting
metabolism is a critical regulator of N- PDGFRα endocytosis
glycan branching by controlling availability
of the sugar-nucleotide UDP-GlcNAc, the We examined oligodendrogenesis in vitro
substrate used by the Mgat family of N- using mouse neural stem cells (NSC)
glycan branching enzymes (19,20,24,37,38). derived from the medial ganglionic
UDP-GlcNAc is generated in the eminence of E12.5 mouse embryos, where
hexosamine pathway de novo from glucose OPC’s first appear. GlcNAc treatment of
or by salvage from N-acetylglucosamine NSCs for 48hrs in growth media lacking
(GlcNAc). Extracellular GlcNAc enters exogenous differentiation cytokines (i.e. no
cells through micropinocytosis, with PDGF-AA, T3, CNTF) significantly
supplementation of cells or mice with increased N-glycan branching and PDGFRα
GlcNAc inhibiting pro-inflammatory T-cell surface expression (Fig. 1A,B), the former
responses and murine models of assessed by L-PHA (Phaseolus vulgaris
inflammatory demyelination by enhancing leukoagglutinin) flow cytometry (17,22).
N-glycan branching (19,31,37,38). Consistent with increased PDGFRα surface
expression, GlcNAc also promoted pre-
Targeted deletion of galectin-3, a ligand for oligodendrocyte differentiation as evidenced
N-glycan branching, leads to decreased by augmented expression of
production of oligodendrocytes, poor Oligodendrocyte Transcription Factor
myelination of axons and reduced ability to (OLIG2) and increased numbers of O4 and
re-myelinate after injury (39). In humans, GalC positive cells (Fig. 1A-C, S1B,C).
loss of function mutations in PGM3, a gene Double staining for PDGFRα and O4
required to generate branched N-glycans revealed that GlcNAc promoted
from GlcNAc, display reduced branching development of pre-oligodendrocytes
and severe CNS hypomyelination (40). (PDGFRα+O4+) with no change in the
Platelet derived growth Factor – AA plays a number of OPC’s (PDGFRα+O4-) (Fig. 1D).
critical role in oligodendrogenesis (41), with Thus, after only two days of culture,
its receptor (PDGFRα) expressed in GlcNAc initiated oligodendrogenesis from
3NSC’s despite the absence of exogenous Next, we examined whether oral GlcNAc
differentiation cytokines such as PDGF-AA. can cross the blood-brain barrier to promote
Remarkably, GlcNAc in growth media was oligodendrocyte differentiation and
more potent than differentiation media myelination in vivo. Adult mice (n=6) and
containing exogenous PDGF-AA at lactating mothers were provided
13
initiating oligodendrogenesis (Fig. S1D). with/without C -labelled GlcNAc
Combining GlcNAc with PDGF-AA also ([U13C]GlcNAc) (10) in their drinking water
enhanced NSC differentiation to O4+ pre- and metabolites derived from perfused
oligodendrocytes (Fig. S1E). brains were analyzed by Liquid
chromatography - tandem mass
To confirm a role for N-glycan branching in spectroscopy (LC-MS/MS). Although this
oligodendrogenesis, we first used method does not resolve stereoisomers of N-
kifunensine to inhibit N-glycan branching Acetylhexosamines (ie. GlcNAc versus
(23) in NSC induced to differentiate by GalNAc), a reversible 4-epimerase
exogenous PDGF-AA. Reducing branching (GALE) equilibrates UDP-GlcNAc and
in NSC’s using kifunensine significantly UDP-GalNAc in vivo (19). LC-MS/MS
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
reduced PDGFRα surface expression and the identified UDP-[U13C]-N-
number of O4+ cells induced by PDGF-AA Acetylhexosamines (UDP-[U13C]-HexNAc)
differentiation media (Fig. S1F). To confirm in treated adult female mouse brains as well
this result genetically, we utilized Mgat5-/- as in the brains of their suckling pups (Fig.
and doxycycline inducible Mgat1f/f/tetO- 2A). This demonstrates that orally delivered
cre/ROSA-rtTA mice (17,22). Mgat5 GlcNAc is not only able to cross the blood
deletion mildly reduces N-glycan branching brain barrier and be metabolized to UDP-
while Mgat1 deletion completely blocks N- GlcNAc by CNS cells, it is also secreted at
glycan branching (Fig. S1A). In vitro sufficient levels in breast milk to raise UDP-
doxycycline treatment of NSC from GlcNAc in the brains of suckling pups.
Mgat1f/f/tetO-cre/ROSA-rtTA mice readily
induced deletion of Mgat1, as measured by To assess whether oral GlcNAc promotes
loss of L-PHA binding (Fig. S1G). Mgat5 oligodendrogenesis in vivo in the absence of
and Mgat1 deleted NSC decreased surface inflammation, we examined primary
levels of PDGFRα and were markedly myelination in mice during the early peri-
reduced in their ability to differentiate into natal period. We provided GlcNAc or
O4+ pre-oligodendrocytes in response to vehicle to pregnant/lactating female mice
PDGF-AA differentiation media (Fig 1E, from E12.5, post-natal day 3 (P3) or P5
S1G). In Mgat5 heterozygous NSC’s, small through to P8. Indeed, oral GlcNAc
reductions in N-glycan branching also increased N-glycan branching in PDGFRα+
inhibited oligodendrocyte differentiation cells as well as the number of pre-
(Fig. 1E). Thus, subtle changes in N-glycan oligodendrocytes (PDGFRα+O4+), immature
branching can markedly impact oligodendrocytes (PDGFRα-O4+) and
oligodendrocyte differentiation from NSC in mature oligodendrocytes (MBP+), with little
vitro. effect on the number of OPC’s
(PDGFRα+O4-) (Fig. 2B, S2A). The lack of
GlcNAc and N-glycan branching promote change in OPC number is consistent with
primary myelination in mice. our in vitro data (Fig. 1D) and suggests that
GlcNAc promotes OPC self-renewal and/or
NSC differentiation to OPC, resulting in a
4stable number of OPC’s. Consistent with tamoxifen induces deletion of Mgat1 in
increased oligodendrogenesis, oral GlcNAc OPC’s but not mature oligodendrocytes.
also increased primary myelination when Indeed, eight weeks but not two weeks after
provided to pups from P3-8, as assessed by tamoxifen treatment, deletion of Mgat1 in
increased levels of staining for myelin basic OPC’s significantly reduced levels of MBP,
protein (MBP) and myelin (as measured by myelin (fluoromyelin) and number of total
fluoromyelin) (Fig. 2C). (Olig2+) and mature (Olig2+CC1+)
oligodendrocytes along with increased
To confirm that N-glycan branching numbers of immature (Olig2+CC1-)
promotes myelination in the absence of oligodendrocytes (Fig 2E, S2E). Tamoxifen
inflammation in vivo, we generated mice has been reported to promote
with tamoxifen inducible deletion of Mgat1 myelination(47), however Mgat1 deletion
only in OPC’s and oligodendrocytes, namely reduced myelination despite potential
Mgat1f/fPlp1-cre/ERTc+ mice. As proteolipid positive effects of tamoxifen. Together,
protein (PLP) promoter driven Cre these data demonstrate that GlcNAc and N-
expression only becomes restricted to the glycan branching promote primary
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
oligodendrocyte lineage (OPC and myelination in mice by driving OPC
oligodendrocyte) at P28 (43), we focused on differentiation.
adult mice. OPC’s continue to proliferate
and generate significant new myelin in GlcNAc prevents damage to demyelinated
adulthood, with myelination gradually axons by promoting myelin repair.
doubling from ~2 to 10 months (44-46).
Tamoxifen readily induced Mgat1 deletion To explore whether GlcNAc can promote re-
in O4+ oligodendrocytes but not 04— cells in myelination in adult mice following myelin
vivo, as determined by loss of L-PHA injury, we utilized the cuprizone model of
binding by flow cytometry (Fig. S2B). non-immune induced de-myelination/re-
Consistent with slow accumulation of new myelination on Mgat5+/- and wildtype
myelin from OPC’s during adulthood, two C57BL/6 mice. Cuprizone at 0.2% induces
weeks following tamoxifen treatment demyelination in the corpus callosum by 3
(Mgat1 deletion) adult Mgat1f/fPlp1- weeks, with maximum demyelination at 5-6
cre/ERTc+ mice did not display significant weeks. Partial re-myelination via maturation
differences in brain levels of MBP or myelin of OPC’s begins at the height of
(fluoromyelin) relative to tamoxifen treated demyelination and becomes complete ~3-5
controls (Fig. S2C). However, eight weeks weeks after cuprizone withdrawal. Given
after initial tamoxifen treatment, Mgat1 this, we examined four different treatment
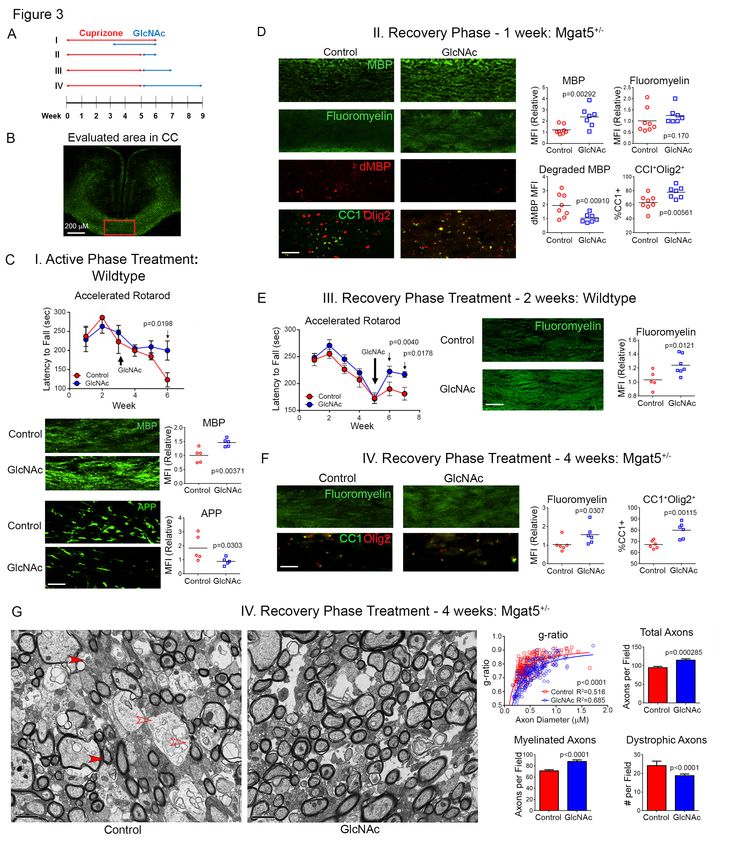
deletion resulted in significant reductions in regimens (Fig. 3A). When GlcNAc was
levels of MBP, myelin (fluoromyelin) and concurrently provided during the final 3
number of total (Olig2+) and mature weeks of a 6-week cuprizone (0.2%)
(Olig2+CC1+) oligodendrocytes along with exposure in wildtype mice, GlcNAc
increased numbers of immature (Olig2+CC1-) prevented loss of motor function (as
oligodendrocytes (Fig. 2D, S2D). To measured using rotarod fall latency) while
confirm that these results primarily arose increasing MBP levels and reducing axonal
from a defect in new myelin formation from damage (as measured by reduced
OPC’s, rather than a defect in mature accumulation of amyloid precursor protein
oligodendrocytes, we generated (APP)) in the corpus callosum (Fig. 3C). To
f/f +
Mgat1 Pdgfra-creER mice where address potential confounding effects of
5GlcNAc on inhibiting demyelination by patients to correlate endogenous serum
cuprizone during concurrent treatment, we HexNAc levels with measures of white
initiated treatment of wildtype mice for 2 matter damage by magnetic resonance
weeks or Mgat5+/- mice for 1 or 4 weeks of imaging (MRI) of the brain. Increased T2w
GlcNAc only after cuprizone was stopped lesion volume and count on brain MRI are
(Fig. 3A). This revealed that GlcNAc measures of the extent and frequency of
enhanced levels of MBP, myelin demyelination, respectively. T2w lesion
(fluoromyelin) and mature oligodendrocytes volume correlated with lower HexNAc
(CC1+Olig2+) while reducing the amount of serum levels (Fig. 4A, p=0.020), whereas
degraded MBP (dMBP)/myelin T2w lesion count did not (p=0.387).
degeneration within the corpus callosum Likewise, patients with contrast enhancing
(Fig. 3D-F). dMBP was detected by an lesions, a marker of active inflammation in
antibody that specifically recognizes areas MS, had similar serum HexNAc levels to
of myelin degeneration (48). Electron those without (p=0.866), suggesting
microscopy analysis confirmed these results, GlcNAc primarily impacts the extent of
revealing that GlcNAc enhanced the number permanent demyelination rather than
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
of myelinated axons, and the degree of initiation of inflammatory demyelination.
myelination as measured by the g-ratio, T1w/T2w ratio maps (50) reflect
while also reducing axon loss and the microstructural integrity of myelin/axons in
number of degenerating and normal appearing white matter (NAWM)
dystrophic/swollen axons (Fig. 3G, S3A). (51) and cortical grey matter(52,53). With
GlcNAc also enhanced the number of age and gender as covariates, low serum
paranodes, which increase with re- HexNAc levels were strongly associated
myelination (Fig. S3A)(49). Enhancement with lower T1w/T2w ratios indicating
of myelination by GlcNAc depends on time, microstructural damage of myelin/axons in
as the increase in fluoromyelin staining in both normal appearing white matter (r2=0.18,
Mgat5+/- mice was ~2-fold greater with 4 p=2.25×10−5) and grey matter (r2=0.23,
versus 1 week of GlcNAc treatment (Fig. p=1.32×10−6), (Fig. 4B,C). Together, these
3D, F). Importantly, the subtle reductions in data are consistent with our mouse data and
N-glycan branching induced in Mgat5+/- suggest that GlcNAc may promote
mice did not alter baseline levels of myelin myelination in MS.
yet reduced re-myelination following
cuprizone induced injury relative to Discussion
Mgat5+/+ control mice (Fig. S3B, C).
Together, these data indicate that GlcNAc Here we report a novel pathway for
and N-glycan branching promotes myelin regulating oligodendrogenesis, primary
repair and provides neuro-protection to myelination and myelin repair by N-glycan
axons following demyelination. branching and GlcNAc. Our data
demonstrates that GlcNAc and N-glycan
A marker of serum GlcNAc inversely branching are neuroprotective for
associates with imaging markers of demyelinated axons by promoting
myelin-axon damage. oligodendrogenesis and myelination from
OPC’s. The association of low endogenous
To explore whether alterations in GlcNAc GlcNAc with increased myelin-axon
may impact myelination status in MS microstructural damage in MS patients
patients, we used a cohort of 180 MS suggests this mechanism is relevant to
6pathogenesis of MS. This hypothesis is drive myelination and promote axonal health.
consistent with recent data suggesting that For example, cell motility is significantly
some MS patients are blocked in their ability enhanced by N-glycan branching via
to generate new myelin from progenitors (9). reduced clustering of integrins (58,59).
The mechanisms that drive Such activity in OPC’s would enhance their
neurodegeneration in MS are poorly ability to traffic to sites of demyelination
understood and our data raise the possibility and promote myelin repair. N-glycan
that alterations in N-glycan branching and/or branching also stimulates glucose
GlcNAc availability may promote transporter surface retention, to enhance
neurodegeneration by blocking glucose uptake (19,25). Glucose-transporter-
remyelination. Indeed, we find that low 1 (GLUT1) in oligodendrocytes promotes
levels of serum GlcNAc in MS patients is axonal health and function by increasing
associated with a progressive disease course, transfer of lactate to axons via increased
clinical disability and multiple neuroimaging glucose supply to the glycolytic pathway in
measures of neurodegeneration (unpublished oligodendrocytes (60). Thus, part of the
data). neuroprotective effect of GlcNAc following
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
myelin repair may be through enhanced
Providing oral GlcNAc to lactating female transport of glucose into oligodendrocytes.
mice increased primary myelination in
nursing pups via delivery of GlcNAc in GlcNAc and/or N-glycan branching also
breast milk. In humans, GlcNAc is a major play important roles in suppressing T cell
component of breast milk oligosaccharides and B cell mediated inflammatory
(~1.5 to ~0.6mg/ml from term to 13 demyelination (17,28,30-33,35,37,38). . In
weeks)(54), and can be released as a T cells, GlcNAc and N-glycan branching
monosaccharide by infant microbiota (55). suppress activation signaling via the T cell
Thus, breast fed newborns consume ~0.5 - receptor (17,22,26), inhibit pro-
1.5g of GlcNAc per day or ~100- inflammatory TH1 and TH17 differentiation
300mg/kg/day for a 5kg infant. This is (24,38) and enhance anti-inflammatory T
similar to the ~160mg/kg/day dose that we regulatory cell differentiation (24). B cell
observed to promote myelination in adult depletion is a potent therapy in MS,
mice. In contrast to human breast milk, predominantly acting by suppressing innate
GlcNAc is not a significant component of antigen presenting cell function rather than
commercial baby formula. Breast-fed via antibody production (61,62). N-glycan
infants display increased myelination and branching in B cells reduces pro-
cognitive function compared to formula fed inflammatory innate signaling via toll-like
infants (56,57), but the mechanism is receptors and inhibits antigen presenting cell
unknown. Our data suggests that GlcNAc in activity, yet promotes adaptive responses
human breast-milk may be a major driver of through the B cell receptor(28). Thus, oral
this effect. GlcNAc is uniquely positioned as a
therapeutic to reverse three major targets
GlcNAc and N-glycan branching markedly driving MS pathogenesis, namely pro-
enhanced cell surface expression of inflammatory T cell responses, pro-
PDGFRα, a critical initiator of OPC inflammatory innate B cell activity and
differentiation. However, GlcNAc and N- myelin repair. No current MS therapy has
glycan branching likely impact other cell such diverse mechanisms of action.
surface receptors/transporters in OPC’s to
7Concentrations of GlcNAc required to raise Mice were bred and utilized as approved by
N-glycan branching in vivo are markedly the University of California, Irvine
lower than those required for in vitro Institutional Animal Care and Use
activity (37,38). This is largely driven by Committee. Dorsal forebrain cortical tissue
GlcNAc entering cells by macropinocytosis, was dissected from the medial ganglionic
and therefore both time and rate of eminence (MGE) at embryonic day 12.5
membrane turnover can influence (E12) of CD1 mice (Charles River) or
concentrations of GlcNAc required to raise Mgat5-/- C57BL/6 mice and their wildtype
N-glycan branching (37,38). Thus, short- littermates and placed in dissection buffer:
term in vitro experiments require high PBS, 0.6% glucose, 50 U/mL Pen/Strep.
concentrations to raise intracellular GlcNAc Tissue from multiple embryos within the
levels quickly while primary cells remain same litter were pooled, and a subsequent
viable. In contrast, cells can be exposed to culture from a single litter was considered a
GlcNAc over a longer time period in vivo, biological repeat. The tissue was dissociated
allowing lower concentrations to be using 0.05% Trypsin-EDTA at 37o C for 10
effective at raising N-glycan branching. The min, followed by treatment with soybean
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
rate of macropinocytosis may also be much trypsin inhibitor (Life Technologies).
higher in vivo compared to in vitro. Dissociated cells were re-suspended in
proliferation medium containing DMEM, 1x
GlcNAc is also known to be highly safe in B27, 1x N2, 1 mM sodium pyruvate, 2 mM
humans. In addition to breastfed infants L-glutamine, 1 mM N-acetylcysteine, 20
consuming significant quantities, large ng/mL EGF (PeproTech), 10 ng/mL bFGF
intravenous doses of GlcNAc (20g and 100g) (PeproTech), and 2 µg/mL heparin and
in humans demonstrated no toxicity issues seeded at 150,000 cells/mL (non-tissue
and no alterations in blood glucose or culture treated plastic plates) and grown as
insulin (63,64). Moreover, treatment with non-adherent spheres. Cell cultures were
insulin had no effect on the serum half-life passaged approximately every 3 days using
of GlcNAc (63). Oral GlcNAc (3-6g/day) enzyme-free NeuroCult Chemical
has also been used in 12 children with Dissociation Kit (Mouse) (StemCell
inflammatory bowel disease for ~2 years Technologies). Cultures were passaged at
without reported toxicities and/or side least once prior to experimental use. For
effects (65). In rats, chronic systematic experiments, passaged cells were cultured in
toxicological studies at doses of 2323- proliferation media (bFGF and EGF) or
2545mg/kg/day for up to 114 weeks found differentiation media (bFGF (10ng/ml) and
no toxicity (66,67). Coupled with PDGF-AA (10ng/ml); Life technologies) for
availability as a dietary supplement, oral 48 hours with or without the presence of
GlcNAc may provide a potent, inexpensive GlcNAc (Ultimate Glucosamine, Wellesley
and safe therapy for MS. Large double-blind Therapeutics) or kifunensine (GlycoSyn).
placebo-controlled trials are warranted to Neurospheres were dispersed using the
investigate this hypothesis. enzyme-free NeuroCult kit before being
analyzed by flow cytometry using one or
Experimental procedures more of the following antibodies: anti-
CD140a/PDGF-RA PE conjugate (1:200,
Mouse brain and neural stem cell A15785, Molecular Probes), anti-O4 Alexa
isolation and analysis Fluor 488 conjugated (1:200, FAB1326G,
RnD systems), Anti-GalC Alexa Fluor 647
8(1:200, MAB342-AF647, Millipore), Anti- of 20mg/mL. Mgat1f/fPlp1-cre/ERTc+ and
Olig2 (1:200, AB9610, Millipore) with anti- Mgat1f/fPdgfra-creER+ (mean age: P71.24,
rabbit Alexa Fluor 488 (1:200, thermofisher). std. dev. 1.393) mice and their control
GlcNAc treatment of mouse pups - GlcNAc Mgat1f/f littermates were injected
(1mg/mL) in drinking water was provided to intraperitoneally with tamoxifen (75mg/kg)
pregnant PLJ mothers or mothers who daily for 3 days starting on day 0 and
recently delivered pups and were nursing sacrificed at two weeks or re-treated with
their young. After the treatment period, pups tamoxifen and sacrificed at 8 weeks. Mice
were anestheszied with isofluorane and were sacrificed following anesthesia and
cardiac perfused with PBS. Pup and fetal cardiac perfusion with phosphate buffered
brains were removed and homogenized by saline. Brains examined by flow cytometry
trituration using glass pipettes in PBS with were first homogenized by trituration using
5% Fetal Bovine Serum (FBS). Cells were glass pipettes in PBS with 5% FBS. Brains
then stained with antibodies and analyzed by examined for myelin content were drop
flow cytometry using antibodies described fixed in 4% paraformaldehyde overnight.
above. For immunofluorescence analysis of
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
pup brains, pups were quickly decapitated, Cuprizone induced demyelination
brains harvested and fixed in 4%
paraformaldehyde overnight. Cuprizone at 0.2% induces demyelination in
the corpus callosum by 3 weeks, with
[U13C]GlcNAc treatment of mice maximum demyelination at 5-6 weeks(68).
8-week-old C57BL/6 mice purchased from
[U13C] GlcNAc was purchased from Jackson Laboratories or 8-week old Mgat5+/-
Omicron Biochemicals and put in the C57BL/6 mice were treated with 0.2%
drinking water at 1mg/mL of female mice Cuprizone (Sigma) mixed into milled rodent
aged 8 weeks for 3 days. Fresh solution of chow for 6 weeks for the active phase
[U13C] GlcNAc in drinking water was treatment and 5 weeks for the recovery
provided each day. After 3 days, mice were treatment. During active phase treatment,
anesthetized with isofluorane and underwent GlcNAc (1mg/mL) in drinking water or just
cardiac perfusion with 50mL of PBS. Brains drinking water (control) was provided for
were harvested and snap frozen in liquid the last 3 weeks of Cuprizone treatment. For
nitrogen. Tissues were cut into 0.04g pieces the recovery phase treatment, GlcNAc in
and crushed mechanically before undergoing drinking water or control was provided after
extraction as described below (Targeted LC- Cuprizone treatment had been stopped.
MS/MS). Levels of UDP-[U13C]GlcNAc Mice were anesthetized and underwent
were measured by LC–MS/MS analysis as cardiac perfusion with 4%
described below (Targeted LC-MS/MS). paraformaldehyde in PBS or 4%
paraformaldehyde plus 0.5% glutaraldehyde
Tamoxifen induced deletion of Mgat1 in Sodium cacodylate buffer for
immunofluorescence or electron
Mgat1f/fPlp1-cre/ERTc+ and Mgat1f/fPdgfra- microscopic analysis respectively. Brains
creER+ were generated by crossing our were then fixed overnight in perfusion
Mgat1f/f mice with Plp1-cre/ERTc+ and solution.
Pdgfra-creER+ lines from Jackson’s
Laboratory. Tamoxifen was dissolved in Accelerated Rotarod
corn oil overnight at 37°C at a concentration
9One day prior to Cuprizone treatment, mice anti-rat Alexa-fluor 488 (1:200,
were trained on the rotarod by allowing Thermofisher), goat anti-rabbit TxRd (1:200,
them to run three 5-minute trials at a Thermofisher). APP is a marker for neuro-
constant 30 rotations per minute (RPM). axonal damage(69-71) while co-staining for
Mice then underwent weekly testing during CC1 and Olig2 are markers of mature
Cuprizone and GlcNAc treatment on an oligodendrocytes(72,73). In order to
accelerating rotarod starting at 4 rpm examine the amount of myelin, slices were
increasing to 40 rpm over 5 minutes. incubated in Fluoromyelin (1:300; F34651,
Latency for mice to fall was recorded. If a Thermofisher) for 45 minutes. Images were
mouse was not running on the rotarod by acquired on a Keyence fluorescence
holding on for 3 turns, this was considered a microscope. Mean fluorescence intensity of
fall. For the active phase treatment, one trial the medial corpus callosum was measured
was run every week. For the recovery phase using ImageJ.
treatment, 3 trials were run for each mouse
each week and latencies were averaged. As Electron Microscopy
expected with Cuprizone treatment
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
performance degraded as treatment Three mice from each treatment group
progressed. Mice whose performance did (control and GlcNAc) were selected
not drop below a pre-determined threshold randomly for EM analysis (before other
(200 seconds) were not used in analysis. investigations were performed). Portions of
these brains from 0 to -1 bregma were rinsed
Immunofluorescence analysis in 0.1 M cacodylate buffer overnight and
again for 15 min. the next day. 2 x 1 mm
For NSC immunoflouresence, whole blocks of the corpus callosum were
neurospheres were seeded onto laminin- dissected out and post-fixed with 1 %
coated coverslips (Neuvitro) in proliferation osmium tetroxide in 0.1 M cacodylate buffer
medium. After 24 hours, proliferation media for 1 hour, rinsed in ddH20, dehydrated in
was removed and replaced with increasing serial dilutions of ethanol (70%,
differentiation medium (same components 85%, 95%, 100% × 2) for 10 min each, put
as proliferation medium but excluding EGF, in propylene oxide (intermediate solvent) for
bFGF, and heparin) to induce differentiation. 2 x 10 min, incubated in propylene
For analysis of mouse brains, brains were oxide/Spurr's resin (1:1 mix) for 1 h, and
incubated in 30% sucrose for at least 72 then in Spurr's resin overnight. The blocks
hours, embedded in OCT (Tissue-tek), were put in a fresh change of resin in flat
frozen for at least 48 hours at -80οC, and embedding molds the next day and
then cut at 40 microns on a cryostat. polymerized overnight at 60 ° Celsius. The
Multiple sections from -1 bregma to -2.5 blocks were sectioned at 1 µm using a Leica
bregma were then stained with antibodies Ultracut UCT ultramicrotome. Floating
for MBP (1:100; MAB386, Millipore), sections were stained in toluidine blue
Olig2 (1:200; AB9610, Millipore), CC1 (1 % toluidine blue and 2 % sodium borate
(1:100; OP90, Millipore), degraded MBP(48) in ddH20) at 60 ° C for 3 min., mounted on
(1:200, AB5864, Millipore) and Amyloid slides and cover-slipped. Ultrathin sections
Precursor Protein (APP, 1:200; clone were sectioned at 70 nm using a Leica
22C11). After overnight incubation with Ultracut UCT ultramicrotome. Sections
primary antibody, tissues were washed and were mounted on 150 mesh copper grids,
incubated with secondary antibodies: goat stained with uranyl acetate and lead citrate
10and viewed using a JEOL 1400 electron MRI was performed at 1.5 Tesla using three-
microscope. Images were captured using a dimensional T1-weighted magnetization
Gatan digital camera. A blinded rater prepared rapid acquisition and multiple
analyzed images by calculating the g-ratio gradient echo sequences (MPRAGE; T1w)
(ratio of the diameter of the axon excluding and axial T2-weighted (T2w) sequences.
the myelin sheath divided by the axon Images were either acquired on a Sonata
diameter including the myelin sheath) as MRI (Siemens Medical Systems, Erlangen,
well as counting the number of total axons, Germany) with TE 4.38 ms, TR 2,110 ms,
myelinated axons, dystrophic axons (defined TI 1.1 ms, flip angle 15° and isotropic
as axon diameter >0.7µm), degenerating resolutions 1 mm3 for T1w, and Multiecho
axons and paranodes. Degenerating axons TSE with TE 81 ms, TR 5,780 ms, 150° flip
were identified as axonal swellings angle, resolution 0.5x0.5x3 mm, no gap for
containing more than 5 clustered dark T2w, or on an Avanto MRI (Siemens
mitochondria and lysosomes. Paranodes Medical Systems, Erlangen, Germany) with
were identified as axons with close TE 3.09 ms, TR 1,900 ms, TI 1.1 ms, flip
proximity of the axolemma with the inner angle 15° and isotropic resolutions 1 mm3
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
membrane of the myelin sheath with a for T1w, and 3D TSE with TE 175 ms, TR
surrounding cytoplasmic portion of 3,000 ms, flip angle 120°, isotropic
oligodendrocyte. resolutions 1 mm3 for T2w. Conventional
spin-echo T1-weighted images (TR 1060 ms,
MS patient cohort TE 14 ms, 3 mm slice thickness, no gap and
44 contiguous axial slices) were obtained
MS patients were recruited from the before and 5 minutes after injection of 0.1
neuroimmunology clinical trial unit at the mmol/kg Gd-DTPA (Magnevist, Bayer-
NeuroCure Clinical Research Center, Schering, Berlin, Germany).
Charité - Universitätsmedizin Berlin (Table
S1). Inclusion criteria were MS based on the T2w lesion segmentation was performed as
2010 revised McDonald criteria2, stable previously described(75) using a semi-
immunomodulatory therapy (RRMS) or no automated procedure including image co-
treatment (PPMS and SPMS). Exclusion registration using FLIRT (FMRIB Software
criteria were acute relapse and/or Library, Oxford, UK) and inhomogeneity
corticosteroids within 6 months prior to correction as embedded into the MedX
inclusion. Disease course was determined v3.4.3 software package (Sensor Systems
under strict adherence to the 1996 Lublin Inc., Sterling, VA, USA). Bulk white matter
criteria(74). Blood draws were fasting. The lesion load and lesion count of T2w scans
study was approved by the local ethics were routinely measured using MedX.
committee of Berlin (Landesamt für
Gesundheit und Soziales (LAGeSo)). All For calculation of T1w/T2w ratio maps,
study participants gave written informed MPRAGE, FLAIR and T2w scans were
consent. Studies were conducted in reoriented to standard space, bias field
conformity with the 1964 Declaration of corrected and cropped to a robust field of
Helsinki in its currently applicable version. view using FSL 5.0.9(76). The MPRAGE
and FLAIR scans were then linearly co-
Magnetic Resonance Imaging (MRI) registered to T2w using FSL FLIRT and
then registered to MNI (Montreal
Neurological Institute) space and brain
11extracted using the Brain Extraction spectrometer (AB Sciex 4000Qtrap, Toronto,
Toolbox (BET)(76). T2w lesions were then ON, Canada). MultiQuant software (AB
automatically segmented by applying the Sciex, Version 2.1) was used for peak
lesion prediction algorithm (LPA) to FLAIR analysis and manual peak confirmation. The
scans, implemented in the Lesion results, expressed as area ratio (area of
Segmentation Toolbox version 2.0.15(77) analyte/area of internal standard), were
for SPM. GM, WM and brain masks were exported to Excel, and analyzed with
then extracted from the MPRAGE. The MetaboAnalyst 3.0. Standard curves were
lesion mask was subtracted from these prepared by adding increasing
masks to remove any lesion effects. The concentrations of GlcNAc or N-Acetyl-D-
T1w/T2w ratio was created by dividing the [UL-13C6]glucosamine ([UL13C6] GlcNAc)
processed MPRAGE scans by the processed (Omicron Biochemicals, Indiana) to 50 µl
T2w scans. Median T1w/T2w ratios were aliquot of control serum (Fig. S4A). This
extracted from the normal appearing WM, way we were able to create a calibration
GM and brain masks. curve for HexNAc serum levels, obtaining
absolute values rather than relative
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
Targeted LC-MS/MS concentrations (Fig. S4B). Analysts were
blinded in regard to sample origin (HC or
Serum samples for metabolomics analysis MS).
were prepared as described previously(78).
Briefly, 50 µL serum (stored at -80oC) and Statistical Analysis
200 µl ice cold extraction solvent (40%
acetonitrile: 40% methanol: 20% H2O), were Statistical analyses for the in vitro and
vortexed for 2 minutes, then shaken in an animal experiments were done with
Eppendorf shaker (Thermomixer R) at 1400 Graphpad Prism by t-tests, ANOVA with
rpm, 4°C for 1 hour and centrifuged at 4°C Sidak’s post-test correction or comparing
for 10 minutes at ~18,000 x g in an best fit curves from non-linear regression
Eppendorf microfuge. Supernatants were (Y=Bmax*X/(Kd + X) as described in the
transferred to a clean tube and evaporated in relevant figure legends. Statistical analyses
a Speedvac (Acid-Resistant CentriVap for the clinical part were performed with R
Vacuum Concentrators, Labconco). Dried Project version 3.5.3. Correlation between
samples were stored at -80°C. Samples were serum HexNAc levels and lesion
resuspended in 100 µl of water containing measurements were analyzed using non-
the Internal Standards D7-Glucose at 0.2 parametric Spearman’s Rho analyses.
mg/mL and H-Tyrosine at 0.02 Correlations between HexNAc levels and
mg/ml. Samples were resolved by LC- T1w/T2w-ration measurements were
MS/MS, in negative mode at the optimum analyzed using linear regression models
polarity in MRM mode on an electrospray with HexNAc levels as an independent
ionization (ESI) triple-quadrupole mass variable.
Data Availability
All data are contained within the manuscript.
Acknowledgements
12We thank Cynthia Kraut and Susan Pikol for MRI assistance and Bibiane Seeger and Carey F. Li
for laboratory assistance. We thank Oswald Steward and Ilse Sears-Kraxberger for providing
electron microscopy services.
Author Contributions
MS (Sy) conceived and coordinated the study with AUB and MD, performed the in vitro and in
vivo animal experiments along with SUL, and wrote the manuscript together with MD and AUB.
AUB conceived and coordinated the study with MD and MS, participated in clinical data
collection, contributed to image analysis and statistical analysis, and wrote the manuscript
together with MS and MD.
SUL maintained animal colonies and performed in vivo animal experiments
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
BLN assisted with study coordination.
JP performed mass spectroscopy.
AG helped perform in vitro experiments and was the blinded rater for in vivo experiments
AAR developed the LC-MS/MS analysis to detect serum GlcNAc.
ZY performed statistical planning of the interventional study and contributed to statistical
analysis of the whole study.
GC calculated the T1w/T2w ratio measurements.
MS (Scheel) coordinated MRI measurements and setup MRI protocols
FP planned and supervised the clinical study.
JWD planned and coordinated mass spectroscopy measurements and analysis.
MD conceived and coordinated the study with AUB and MS, coordinated the serum analysis,
supervised the mouse studies and wrote the manuscript together with MS and AUB.
Funding
Research was supported by a Marilyn Hilton innovator award to MS as well as R01AT007452
and R01AI144403 to MD. The MS cohort and MRI study was supported by DFG grant Exc. 257
to FP. The content is solely the responsibility of the authors and does not necessarily represent
the official views of the National Institutes of Health.
Conflict of Interest
13AUB, FP, JD and MD are named as inventors on a patent that describes GlcNAc as a biomarker
for multiple sclerosis. JD and MD are named as inventors on a patent for use of GlcNAc in MS
and were co-founders of Glixis Therapeutics, a company developing analogs of GlcNAc for MS
and other autoimmune diseases.
References
1. Reich, D. S., Lucchinetti, C. F., and Calabresi, P. A. (2018) Multiple Sclerosis. N Engl J
Med 378, 169-180
2. Fields, R. D. (2008) White matter in learning, cognition and psychiatric disorders. Trends
Neurosci 31, 361-370
3. Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., Liu, Y.,
Tsingalia, A., Jin, L., Zhang, P. W., Pellerin, L., Magistretti, P. J., and Rothstein, J. D.
(2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration.
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
Nature 487, 443-448
4. Fünfschilling, U., Supplie, L. M., Mahad, D., Boretius, S., Saab, A. S., Edgar, J.,
Brinkmann, B. G., Kassmann, C. M., Tzvetanova, I. D., Möbius, W., Diaz, F., Meijer, D.,
Suter, U., Hamprecht, B., Sereda, M. W., Moraes, C. T., Frahm, J., Goebbels, S., and
Nave, K. A. (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal
integrity. Nature 485, 517-521
5. Mei, F., Lehmann-Horn, K., Shen, Y. A., Rankin, K. A., Stebbins, K. J., Lorrain, D. S.,
Pekarek, K., S, A. S., Xiao, L., Teuscher, C., von Budingen, H. C., Wess, J., Lawrence, J.
J., Green, A. J., Fancy, S. P., Zamvil, S. S., and Chan, J. R. (2016) Accelerated
remyelination during inflammatory demyelination prevents axonal loss and improves
functional recovery. eLife 5
6. Patrikios, P., Stadelmann, C., Kutzelnigg, A., Rauschka, H., Schmidbauer, M., Laursen,
H., Sorensen, P. S., Brück, W., Lucchinetti, C., and Lassmann, H. (2006) Remyelination
is extensive in a subset of multiple sclerosis patients. Brain 129, 3165-3172
7. Goldschmidt, T., Antel, J., König, F. B., Brück, W., and Kuhlmann, T. (2009)
Remyelination capacity of the MS brain decreases with disease chronicity. Neurology 72,
1914-1921
8. Patani, R., Balaratnam, M., Vora, A., and Reynolds, R. (2007) Remyelination can be
extensive in multiple sclerosis despite a long disease course. Neuropathology and applied
neurobiology 33, 277-287
9. Yeung, M. S. Y., Djelloul, M., Steiner, E., Bernard, S., Salehpour, M., Possnert, G.,
Brundin, L., and Frisen, J. (2019) Dynamics of oligodendrocyte generation in multiple
sclerosis. Nature 566, 538-542
10. Jakel, S., Agirre, E., Mendanha Falcao, A., van Bruggen, D., Lee, K. W., Knuesel, I.,
Malhotra, D., Ffrench-Constant, C., Williams, A., and Castelo-Branco, G. (2019) Altered
human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543-547
11. Galloway, D. A., Gowing, E., Setayeshgar, S., and Kothary, R. (2020) Inhibitory milieu
at the multiple sclerosis lesion site and the challenges for remyelination. Glia 68, 859-877
12. Mi, S., Hu, B., Hahm, K., Luo, Y., Kam Hui, E. S., Yuan, Q., Wong, W. M., Wang, L.,
Su, H., Chu, T. H., Guo, J., Zhang, W., So, K. F., Pepinsky, B., Shao, Z., Graff, C.,
14Garber, E., Jung, V., Wu, E. X., and Wu, W. (2007) LINGO-1 antagonist promotes spinal
cord remyelination and axonal integrity in MOG-induced experimental autoimmune
encephalomyelitis. Nature medicine 13, 1228-1233
13. Stoffels, J. M., de Jonge, J. C., Stancic, M., Nomden, A., van Strien, M. E., Ma, D.,
Siskova, Z., Maier, O., Ffrench-Constant, C., Franklin, R. J., Hoekstra, D., Zhao, C., and
Baron, W. (2013) Fibronectin aggregation in multiple sclerosis lesions impairs
remyelination. Brain 136, 116-131
14. Sloane, J. A., Batt, C., Ma, Y., Harris, Z. M., Trapp, B., and Vartanian, T. (2010)
Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through
TLR2. Proceedings of the National Academy of Sciences of the United States of America
107, 11555-11560
15. Kotter, M. R., Li, W. W., Zhao, C., and Franklin, R. J. (2006) Myelin impairs CNS
remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci 26,
328-332
16. Mi, S., Miller, R. H., Tang, W., Lee, X., Hu, B., Wu, W., Zhang, Y., Shields, C. B.,
Zhang, Y., Miklasz, S., Shea, D., Mason, J., Franklin, R. J., Ji, B., Shao, Z., Chedotal, A.,
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
Bernard, F., Roulois, A., Xu, J., Jung, V., and Pepinsky, B. (2009) Promotion of central
nervous system remyelination by induced differentiation of oligodendrocyte precursor
cells. Ann Neurol 65, 304-315
17. Demetriou, M., Granovsky, M., Quaggin, S., and Dennis, J. W. (2001) Negative
regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 409,
733-739
18. Partridge, E. A., Le Roy, C., Di Guglielmo, G. M., Pawling, J., Cheung, P., Granovsky,
M., Nabi, I. R., Wrana, J. L., and Dennis, J. W. (2004) Regulation of cytokine receptors
by Golgi N-glycan processing and endocytosis. Science 306, 120-124
19. Lau, K. S., Partridge, E. A., Grigorian, A., Silvescu, C. I., Reinhold, V. N., Demetriou,
M., and Dennis, J. W. (2007) Complex N-glycan number and degree of branching
cooperate to regulate cell proliferation and differentiation. Cell 129, 123-134
20. Dennis, J. W., Nabi, I. R., and Demetriou, M. (2009) Metabolism, cell surface
organization, and disease. Cell 139, 1229-1241
21. Chen, I. J., Chen, H. L., and Demetriou, M. (2007) Lateral compartmentalization of T cell
receptor versus CD45 by galectin-N-glycan binding and microfilaments coordinate basal
and activation signaling. J Biol Chem 282, 35361-35372
22. Zhou, R. W., Mkhikian, H., Grigorian, A., Hong, A., Chen, D., Arakelyan, A., and
Demetriou, M. (2014) N-glycosylation bidirectionally extends the boundaries of
thymocyte positive selection by decoupling Lck from Ca(2)(+) signaling. Nature
immunology 15, 1038-1045
23. Mkhikian, H., Mortales, C. L., Zhou, R. W., Khachikyan, K., Wu, G., Haslam, S. M.,
Kavarian, P., Dell, A., and Demetriou, M. (2016) Golgi self-correction generates
bioequivalent glycans to preserve cellular homeostasis. eLife 5
24. Araujo, L., Khim, P., Mkhikian, H., Mortales, C. L., and Demetriou, M. (2017)
Glycolysis and glutaminolysis cooperatively control T cell function by limiting
metabolite supply to N-glycosylation. eLife 6
25. Ohtsubo, K., Takamatsu, S., Minowa, M. T., Yoshida, A., Takeuchi, M., and Marth, J. D.
(2005) Dietary and genetic control of glucose transporter 2 glycosylation promotes
insulin secretion in suppressing diabetes. Cell 123, 1307-1321
1526. Chen, I. J., Chen, H. L., and Demetriou, M. (2007) Lateral compartmentalization of T cell
receptor versus CD45 by galectin-N-glycan binding and microfilaments coordinate basal
and activation signaling. J Biol Chem 282, 35361-35372
27. Mortales, C. L., Lee, S. U., and Demetriou, M. (2020) N-Glycan Branching Is Required
for Development of Mature B Cells. J Immunol 205, 630-636
28. Mortales, C. L., Lee, S. U., Manousadjian, A., Hayama, K. L., and Demetriou, M. (2020)
N-Glycan Branching Decouples B Cell Innate and Adaptive Immunity to Control
Inflammatory Demyelination. iScience 23, 101380
29. Bahaie, N. S., Kang, B. N., Frenzel, E. M., Hosseinkhani, M. R., Ge, X. N., Greenberg,
Y., Ha, S. G., Demetriou, M., Rao, S. P., and Sriramarao, P. (2011) N-Glycans
differentially regulate eosinophil and neutrophil recruitment during allergic airway
inflammation. J Biol Chem 286, 38231-38241
30. Lee, S. U., Grigorian, A., Pawling, J., Chen, I. J., Gao, G., Mozaffar, T., McKerlie, C.,
and Demetriou, M. (2007) N-glycan processing deficiency promotes spontaneous
inflammatory demyelination and neurodegeneration. J Biol Chem 282, 33725-33734
31. Mkhikian, H., Grigorian, A., Li, C. F., Chen, H. L., Newton, B., Zhou, R. W., Beeton, C.,
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
Torossian, S., Tatarian, G. G., Lee, S. U., Lau, K., Walker, E., Siminovitch, K. A.,
Chandy, K. G., Yu, Z., Dennis, J. W., and Demetriou, M. (2011) Genetics and the
environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat Commun
2, 334
32. Li, C. F., Zhou, R. W., Mkhikian, H., Newton, B. L., Yu, Z., and Demetriou, M. (2013)
Hypomorphic MGAT5 polymorphisms promote multiple sclerosis cooperatively with
MGAT1 and interleukin-2 and 7 receptor variants. Journal of neuroimmunology
33. Brynedal, B., Wojcik, J., Esposito, F., Debailleul, V., Yaouanq, J., Martinelli-Boneschi,
F., Edan, G., Comi, G., Hillert, J., and Abderrahim, H. (2010) MGAT5 alters the severity
of multiple sclerosis. J Neuroimmunol 220, 120-124
34. Ye, Z., and Marth, J. D. (2004) N-glycan branching requirement in neuronal and
postnatal viability. Glycobiology 14, 547-558
35. Grigorian, A., and Demetriou, M. (2011) Mgat5 deficiency in T cells and experimental
autoimmune encephalomyelitis. ISRN Neurol 2011, 374314
36. Backer-Koduah, P., Infante-Duarte, C., Ivaldi, F., Uccelli, A., Bellmann-Strobl, J.,
Wernecke, K. D., Sy, M., Demetriou, M., Dorr, J., Paul, F., and Ulrich Brandt, A. (2020)
Effect of vitamin D supplementation on N-glycan branching and cellular
immunophenotypes in MS. Ann Clin Transl Neurol
37. Grigorian, A., Lee, S. U., Tian, W., Chen, I. J., Gao, G., Mendelsohn, R., Dennis, J. W.,
and Demetriou, M. (2007) Control of T Cell-mediated autoimmunity by metabolite flux
to N-glycan biosynthesis. J Biol Chem 282, 20027-20035
38. Grigorian, A., Araujo, L., Naidu, N. N., Place, D., Choudhury, B., and Demetriou, M.
(2011) N-acetylglucosamine inhibits T-helper 1 (Th1) / T-helper 17 (Th17) responses and
treats experimental autoimmune encephalomyelitis. The Journal of biological chemistry
39. Pasquini, L. A., Millet, V., Hoyos, H. C., Giannoni, J. P., Croci, D. O., Marder, M., Liu,
F. T., Rabinovich, G. A., and Pasquini, J. M. (2011) Galectin-3 drives oligodendrocyte
differentiation to control myelin integrity and function. Cell death and differentiation 18,
1746-1756
40. Zhang, Y., Yu, X., Ichikawa, M., Lyons, J. J., Datta, S., Lamborn, I. T., Jing, H., Kim, E.
S., Biancalana, M., Wolfe, L. A., DiMaggio, T., Matthews, H. F., Kranick, S. M., Stone,
16K. D., Holland, S. M., Reich, D. S., Hughes, J. D., Mehmet, H., McElwee, J., Freeman,
A. F., Freeze, H. H., Su, H. C., and Milner, J. D. (2014) Autosomal recessive
phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune
deficiency, autoimmunity, and neurocognitive impairment. The Journal of allergy and
clinical immunology 133, 1400-1409, 1409 e1401-1405
41. Hu, J. G., Fu, S. L., Wang, Y. X., Li, Y., Jiang, X. Y., Wang, X. F., Qiu, M. S., Lu, P. H.,
and Xu, X. M. (2008) Platelet-derived growth factor-AA mediates oligodendrocyte
lineage differentiation through activation of extracellular signal-regulated kinase
signaling pathway. Neuroscience 151, 138-147
42. Rivers, L. E., Young, K. M., Rizzi, M., Jamen, F., Psachoulia, K., Wade, A., Kessaris, N.,
and Richardson, W. D. (2008) PDGFRA/NG2 glia generate myelinating
oligodendrocytes and piriform projection neurons in adult mice. Nature neuroscience 11,
1392-1401
43. Michalski, J. P., Anderson, C., Beauvais, A., De Repentigny, Y., and Kothary, R. (2011)
The proteolipid protein promoter drives expression outside of the oligodendrocyte lineage
during embryonic and early postnatal development. PloS one 6, e19772
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
44. Young, K. M., Psachoulia, K., Tripathi, R. B., Dunn, S. J., Cossell, L., Attwell, D.,
Tohyama, K., and Richardson, W. D. (2013) Oligodendrocyte dynamics in the healthy
adult CNS: evidence for myelin remodeling. Neuron 77, 873-885
45. Sturrock, R. R. (1980) Myelination of the mouse corpus callosum. Neuropathol Appl
Neurobiol 6, 415-420
46. Hughes, E. G., Orthmann-Murphy, J. L., Langseth, A. J., and Bergles, D. E. (2018)
Myelin remodeling through experience-dependent oligodendrogenesis in the adult
somatosensory cortex. Nat Neurosci 21, 696-706
47. Gonzalez, G. A., Hofer, M. P., Syed, Y. A., Amaral, A. I., Rundle, J., Rahman, S., Zhao,
C., and Kotter, M. R. N. (2016) Tamoxifen accelerates the repair of demyelinated lesions
in the central nervous system. Sci Rep 6, 31599
448. Cantoni, C., Bollman, B., Licastro, D., Xie, M., Mikesell, R., Schmidt, R., Yuede, C. M.,
Galimberti, D., Olivecrona, G., Klein, R. S., Cross, A. H., Otero, K., and Piccio, L.
(2015) TREM2 regulates microglial cell activation in response to demyelination in vivo.
Acta neuropathologica 129, 429-447
49. Prineas, J. W., and McDonald, W. I. (1997) Demyelinating diseases, Arnold, London,
United Kingdom
50. Glasser, M. F., and Van Essen, D. C. (2011) Mapping human cortical areas in vivo based
on myelin content as revealed by T1- and T2-weighted MRI. The Journal of neuroscience
: the official journal of the Society for Neuroscience 31, 11597-11616
51. Beer, A., Biberacher, V., Schmidt, P., Righart, R., Buck, D., Berthele, A., Kirschke, J.,
Zimmer, C., Hemmer, B., and Muhlau, M. (2016) Tissue damage within normal
appearing white matter in early multiple sclerosis: assessment by the ratio of T1- and T2-
weighted MR image intensity. Journal of neurology 263, 1495-1502
52. Nakamura, K., Chen, J. T., Ontaneda, D., Fox, R. J., and Trapp, B. D. (2017) T1-/T2-
weighted ratio differs in demyelinated cortex in multiple sclerosis. Annals of neurology
82, 635-639
53. Righart, R., Biberacher, V., Jonkman, L. E., Klaver, R., Schmidt, P., Buck, D., Berthele,
A., Kirschke, J. S., Zimmer, C., Hemmer, B., Geurts, J. J. G., and Muhlau, M. (2017)
17Cortical pathology in multiple sclerosis detected by the T1/T2-weighted ratio from
routine magnetic resonance imaging. Annals of neurology 82, 519-529
54. Miller, J. B., Bull, S., Miller, J., and McVeagh, P. (1994) The oligosaccharide
composition of human milk: temporal and individual variations in monosaccharide
components. J Pediatr Gastroenterol Nutr 19, 371-376
55. Lawson, M. A. E., O'Neill, I. J., Kujawska, M., Gowrinadh Javvadi, S., Wijeyesekera, A.,
Flegg, Z., Chalklen, L., and Hall, L. J. (2020) Breast milk-derived human milk
oligosaccharides promote Bifidobacterium interactions within a single ecosystem. ISME J
14, 635-648
56. Deoni, S. C., Dean, D. C., 3rd, Piryatinsky, I., O'Muircheartaigh, J., Waskiewicz, N.,
Lehman, K., Han, M., and Dirks, H. (2013) Breastfeeding and early white matter
development: A cross-sectional study. Neuroimage 82, 77-86
57. Isaacs, E. B., Fischl, B. R., Quinn, B. T., Chong, W. K., Gadian, D. G., and Lucas, A.
(2010) Impact of breast milk on intelligence quotient, brain size, and white matter
development. Pediatr Res 67, 357-362
58. Granovsky, M., Fata, J., Pawling, J., Muller, W. J., Khokha, R., and Dennis, J. W. (2000)
Downloaded from http://www.jbc.org/ by guest on November 9, 2020
Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nature medicine 6,
306-312
59. Demetriou, M., Nabi, I. R., Coppolino, M., Dedhar, S., and Dennis, J. W. (1995) Reduced
contact-inhibition and substratum adhesion in epithelial cells expressing GlcNAc-
transferase V. The Journal of cell biology 130, 383-392
60. Saab, A. S., Tzvetavona, I. D., Trevisiol, A., Baltan, S., Dibaj, P., Kusch, K., Mobius, W.,
Goetze, B., Jahn, H. M., Huang, W., Steffens, H., Schomburg, E. D., Perez-Samartin, A.,
Perez-Cerda, F., Bakhtiari, D., Matute, C., Lowel, S., Griesinger, C., Hirrlinger, J.,
Kirchhoff, F., and Nave, K. A. (2016) Oligodendroglial NMDA Receptors Regulate
Glucose Import and Axonal Energy Metabolism. Neuron 91, 119-132
61. Bar-Or, A., Fawaz, L., Fan, B., Darlington, P. J., Rieger, A., Ghorayeb, C., Calabresi, P.
A., Waubant, E., Hauser, S. L., Zhang, J., and Smith, C. H. (2010) Abnormal B-cell
cytokine responses a trigger of T-cell-mediated disease in MS? Annals of neurology 67,
452-461
62. Cross, A. H., Stark, J. L., Lauber, J., Ramsbottom, M. J., and Lyons, J. A. (2006)
Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients.
J Neuroimmunol 180, 63-70
63. Levin, R. M., Krieger, N. N., and Winzler, R. J. (1961) Glucmsumine and
acetylglucosamine tolerance in man. The Journal of laboratory and clinical medicine 58,
927-932
64. Gaulden, E. C., and Keating, W. C. (1964) The Effect of Intravenous N-Acetyl-D-
Glucosamine on the Blood and Urine Sugar Concentrations of Normal Subjects.
Metabolism: clinical and experimental 13, 466-472
65. Salvatore, S., Heuschkel, R., Tomlin, S., Davies, S. E., Edwards, S., Walker-Smith, J. A.,
French, I., and Murch, S. H. (2000) A pilot study of N-acetyl glucosamine, a nutritional
substrate for glycosaminoglycan synthesis, in paediatric chronic inflammatory bowel
disease. Aliment Pharmacol Ther 14, 1567-1579
66. Lee, K. Y., Shibutani, M., Takagi, H., Arimura, T., Takigami, S., Uneyama, C., Kato, N.,
and Hirose, M. (2004) Subchronic toxicity study of dietary N-acetylglucosamine in F344
18You can also read

















































