Cellular mechanisms linking cancers to obesity - Cell Stress
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Review
www.cell-stress.com
Cellular mechanisms linking cancers to obesity
Xiao-Zheng Liu1,#, Line Pedersen1,# and Nils Halberg1,*
1Department of Biomedicine, University of Bergen, N-5020 Bergen, Norway.
#Equal contribution.
* Corresponding Author:
Nils Halberg, Department of Biomedicine, University of Bergen, Jonas Lies vei 91, 5020 Bergen, Norway; Phone: +47 5558 6442;
E-mail: nils.halberg@uib.no
ABSTRACT Obesity is epidemiologically linked to 13 forms doi: 10.15698/cst2021.05.248
of cancer. The local and systemic obese environment is Received originally: 27.01.2021
complex and likely affect tumors through multiple avenues. in revised form: 23.03.2021,
Accepted 26.03.2021,
This includes modulation of cancer cell phenotypes and the
Published 12.04.2021.
composition of the tumor microenvironment. A molecular
understanding of how obesity links to cancer holds promise
for identifying candidate genes for targeted therapy for Keywords: obesity, cancer, mouse models, adipokines,
obese cancer patient. Herein, we review both the cell- metabolism, inflammation, extracellular Matrix remodeling.
autonomous and non-cell-autonomous mechanisms linking
obesity and cancer as well as provide an overview of the Abbreviatons:
mouse model systems applied to study this. ARG2 – arginase 2; BMI – body mass index; COX2 –
cyclooxygenase-2; DIO – diet-induced obesity; ECM –
extracellular matrix; FABP – fatty acid binding protein; FACS
– fluorescence-activated cell sorting; FFA – free fatty acid;
HCC – hepatocellular carcinoma; HFD – high-fat diet; IGF –
insuline growth factor; IL – interleukin; LFD – low-fat diet;
MMTV – mouse mammary tumor virus; NASH – non-
alcoholic steatohepatitis; PDAC – pancreatic ductal
adenocarcinoma; PPARγ – peroxisome proliferator-activated
receptor γ; PyMT – polyoma middle T antigen; S1P –
sphingosine-1-phosphate; SCFA – short-chain fatty acid;
TGFα – transforming growth factor-alpha; TNFα – tumor
necrosis factor-α.
INTRODUCTION viewed in [5]). Epidemiological cross-comparisons have
Obesity is defined as an excessive accumulation of adipose revealed that these interactions involve biological specifici-
tissue, which stores energy in the form of triglycerides. This ties. Examples include: i) sex-related correlations – the
mainly occurs when caloric intake exceeds energy expendi- association between obesity and colon cancer are signifi-
ture. Currently, 1.9 billion adults and 38 million children cantly more pronounced in males than in females, ii) spe-
worldwide are categorized as overweight – numbers that cific histological subtypes – obesity is correlated with
have tripled since 1975 [1]. Dating back to the mid-1980’s, esophageal adenocarcinoma, but not esophageal squa-
clinicians reported that obese patients diagnosed with mous cell carcinoma and iii) menopause state – the risk of
breast cancer have poorer survival outcomes [2]. Large breast cancer is associated with obesity in postmenopause,
epidemiological studies have since supported and expand- but not in premenopause [6].
ed on these observations and estimates now suggest that The obese environment is highly complex and compris-
obesity is implicated in 15-20% of cancer-related mortali- es changes in serum levels of multiple circulating factors
ties (reviewed in [3, 4]). This is a consequence of obesity’s such as glucose, leptin, glucagon, adiponectin, glucagon-
association with an increased risk of developing cancers like-peptide-1, insulin, free fatty acids (FFA) and cholester-
(cancer incidence) and with specific obesity-linked biologi- ol; altered sleeping behavior; altered gut microbiome; al-
cal effects on cancer progression in combination. Currently, tered pharmacodynamics of therapeutics, poor wound
obesity has been associated with 13 cancer types (re- healing and post-operative infections, and the develop-
OPEN ACCESS | www.cell-stress.com 55 Cell Stress | MAY 2021 | Vol. 5 No. 5X.-Z. Liu et al. (2021) Obesity and cancer
ment of comorbid diseases (including chronic low-grade portantly, these models often support the biological speci-
inflammation, heart disease, type 2 diabetes and hyperten- ficities of the epidemiological link between obesity and
sion). Such phenotypic complexity is present both systemi- cancer. For example, consistent with human data, obesity
cally and in local tissue microenvironments. From the leads to increased tumor burden and faster disease pro-
emerging molecular understanding of how such complexity gression in oncogenic Kras-driven pancreatic ductal adeno-
in the obese environment links to cancers, we are begin- carcinoma (PDAC), but not in lung cancer [7]. Table 1 pre-
ning to uncover new and fascinating interactions between sents an overview that details how both genetic and DIO
the physiological state of host and tumor behavior. models have been applied to cancer research.
To recapitulate the complex obesogenic environment
and uncover the molecular mechanisms that link obesity Genetic animal models
and cancer the field has so far relied on mouse models. In Amongst genetic models, mice with deficient leptin signal-
this review, we provide an overview of these model sys- ing are the most frequently used [8]. In these models, obe-
tems as well as proposed molecular and cellular processes sity is caused by the lack of leptin (the ob/ob mouse), or by
by which the obese environment affects tumor initiation a mutation in the leptin receptor gene (db/db mouse), both
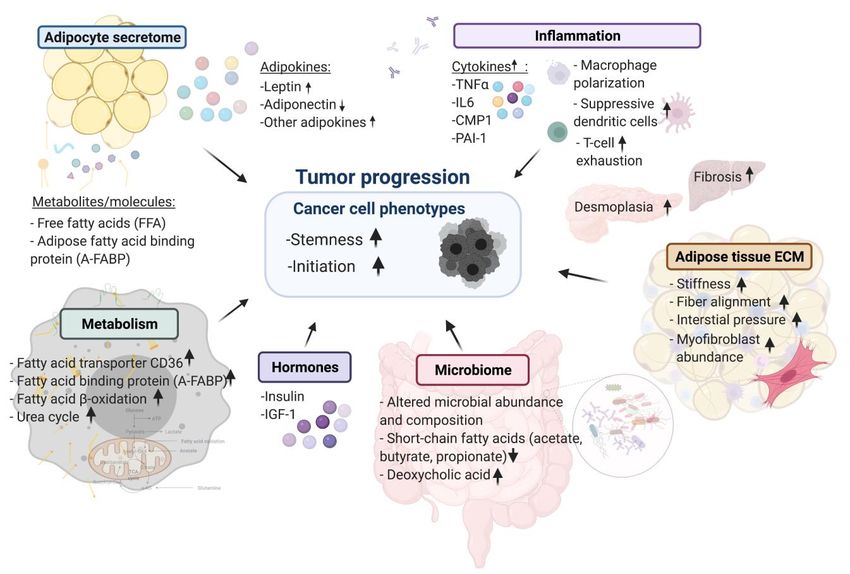
and progression (summarized in Figure 1). causing the mice to overfeed. The genetic models consist-
ently induce an early onset obese state and comorbidities
ANIMAL MODELS OF THE OBESITY-CANCER CONNEC- such as insulin resistance and hepatic steatosis when the
TION mice are fed standard chow. Their primary disadvantage is
As in vitro models generally do not demonstrate the full the exclusion of potential peripheral effects of leptin on
complexity of the obesogenic phenotype, in vivo animal cancer cells and the cells of the tumor microenvironment.
models have been essential research tools to address the
mechanisms underlying the obesity-cancer connection. The DIO animal models
most widely used models are either genetic or diet- The DIO rodent models are established by feeding animals
induced obesity (DIO) models. Upon a cancer challenge with diets containing a high proportion of fat, sugar or a
these models broadly display increased tumor initiation combination of the two. While several feeding patterns
and growth, relative to their non-obese counterparts. Im- have been developed, the most commonly-used diets con-
tain 30-60% kcals from fat which is provided to the mice
FIGURE 1: Summary of cellular mechanisms linking cancers to obesity.
OPEN ACCESS | www.cell-stress.com 56 Cell Stress | MAY 2021 | Vol. 5 No. 5X.-Z. Liu et al. (2021) Obesity and cancer
TABLE 1. Overview of in vitro and in vivo studies.
Obese tumor phenotype/ pro-
Obesity model Diet, duration Cancer model Ref
posed mechanism (keywords)
Breast cancer
HFD (60% kcal from Calorie restriction and rapamycin
T (MMTV-Wnt-1, 5x10^4 cells,
DIO fat), 8 weeks + spe- inhibit mammary tumor growth in [120]
orthotopic)
cial diet postmenopausal obesity
T (E0771/MDA-MB-231, 1×10^6 Obesity-induced expanded adipose
HFD (60% kcal from
DIO cells, orthotopic; LLC/ID8, stromal cells promote tumor [121]
fat)
1x10^6 cells, subcutaneous) growth
Obesity promotes cancer stemness
Western diet (21% phenotype via leptin-STAT3-G9a
DIO CICM (N-methylnitrosourea) [40]
fat), 45 days histone methyltransferase signaling
axis
Obesity-associated NLRC4 inflam-
T (Py8119, 1×10^5 cells /
HFD (36% kcal from masome activation/ interleukin
DIO E0771, 2×10^5 cells, orthoto- [108]
fat), 10 weeks (IL)-1 signalling promotes breast
pic)
tumor growth and angiogenesis
T (86R2 or 99LN, 1.5×10^6 cells, Obesity-associated inflammation
HFD (60% kcal from
DIO orthotopic)/metastasis assay promotes breast cancer metastatic [13]
fat), 15 weeks
(99LN, 2×10^6 cells, tail vein) progression
HFD fed mice have reduced overall
HFD (60% kcal from Metastasis assay (metM-
DIO survival and higher incidence of [122]
fat), 13 weeks Wntlung, 2,5x10^3, tail vein)
lung macrometastases
HFD (60% kcal from A-FABP promotes tumor stemness
T (E0771, 5×10^5 cells + limiting
fat), 5-6 months, and aggressiveness through activa-
DIO dilution, orthotopic), GEMM [90]
GEMM (45% kcal tion of the IL-6/STAT3/ALDH1
(MMTV-TGFα)
from fat), 14 months pathway
Obesity induces hypoxia, neutro-
phil infiltration and EMT, leading to
HFD (60% kcal from
DIO T (E0771 or Py230, orthotopic) the faster growing tumors and an [103]
fat), 9 - 11 weeks
increase in metastasis-initiating
cells
HFD (60% kcal from T (E0771 (1×10^4 - 2×10^5 Metabolically activated adipose
DIO fat), 12 weeks or 4 cells) /PY8119, (2,5 - 5×10^3 tissue macrophages link obesity to [20]
days cells), orthotopic) triple-negative breast cancer
Heparanase regulates macrophage
HFD (60% kcal from T (E0771, 5×10^5 cells, ortho-
DIO functions to promote tumor pro- [86]
fat), 12 weeks topic)
gression
A non-canonical function of BMAL1
HFD (60% kcal from T (E0771, 2 x 10^5 cells, ortho- metabolically limits obesity-
DIO [95]
fat), 5 weeks topic) promoted triple-negative breast
cancer
Enhanced resistin secretion in
HFD (60% kcal from T (E0771, 5×10^5 cells, ortho- obese mammary adipose issue via
DIO [64]
fat), 12 weeks topic) FFA/PPARγ/TAZ axis promote
breast tumorigenesis
Colon/small intestine cancer
HFD (56.7% kcal Adiponectin supresses colorectal
DIO from fat), until end- CICM (azoxymethane) carcinogenesis under the HFD con- [56]
point dition
HFD (56.7% kcal Leptin acts as a growth factor for
DIO, ob/ob,
from fat), until end- CICM (azoxymethane) colorectal tumors at stages sub- [32]
db/db
point suquent to tumor initiation
OPEN ACCESS | www.cell-stress.com 57 Cell Stress | MAY 2021 | Vol. 5 No. 5X.-Z. Liu et al. (2021) Obesity and cancer
TABLE 1 (continued). Overview of in vitro and in vivo studies.
Obese tumor phenotype/ pro-
Obesity model Diet, duration Cancer model Ref
posed mechanism (keywords)
Colon/small intestine cancer
HFD alter expression of inflamma-
HFD (40% kcal from
DIO GEMM (ApcMin/+) tory markers and increase tumor- [123]
fat), 8 weeks
igenesis
HFD (60% kcal from HFD mediated dysbiosis promotes
DIO fat), 22 weeks/until GEMM (KrasG12Dint) carcinogenesis independently of [102]
endpoint obesity
HFD enhances stemness and tu-
HFD (60% kcal from
DIO - morigenicity of intestinal progeni- [12]
fat), 9-14 months
tors
Fatty acid metabolism impair T
HFD (60% kcal from T (MC38, 1×10^5 cells, subcu-
DIO cells infiltration and function and [92]
fat), 8 - 10 weeks taneous)
promote cancer growth
Kidney cancer
HFD promotes dendritic cell infil-
HFD (60% kcal from T (RenCa-Luc, 2 × 10^5 cells, tration, which suppress T cell ex-
DIO [104]
fat), 20 weeks orthotopic) pansion and enhanced tumor
growth
Elevated leptin during diet-Induced
HFD (60% kcal from
DIO, ob/ob T (CRL-2947-Luc, orthotopic) obesity reduces the efficacy of tu- [41]
fat), 20 weeks
mor immunotherapy
Liver cancer
Obesity induced low-grade inflam-
HFD (59% kcal from
DIO, ob/ob CICM (Diethylnitrosamine) mation promoted the hepatic pro- [100]
fat)
carcinogen DEN-induced HCC
CICM (7,12- Obesity-induced gut microbial me-
HFD (60% kcal from
DIO, ob/ob dimethylbenz(a)anthracene, tabolite promotes liver cancer [11]
fat), until endpoint
DMBA) through senescence secretome
ER stress cooperates with hypernu-
HFD (59% kcal from
DIO GEMM (MUP-uPA) trition to trigger TNF-dependent [101]
fat), until endpoint
spontaneous HCC development
Gut microbiota promotes obesity-
CICM (7,12-
HFD (60% kcal from associated liver cancer through
DIO dimethylbenz(a)anthracene, [124]
fat), until endpoint PGE2-mediated suppression of
DMBA)
antitumor immunity
HFD (43% kcal from Obesity drives STAT-3 dependent
DIO Alb-Cre;Ptpn2fl/fl [82]
fat, 40 weeks) hepatocellular carcinoma
Melanoma
Paradoxical effects of obesity on T
HFD (60% kcal from
DIO T (B16,1*10^6, subcutaneous) cell function during tumor progres- [109]
fat)
sion and PD-1 checkpoint blockade
Myeloma
Vk12598 (5x10^5, intrafemoral- Acetyl-CoA synthetase 2 - a critical
HFD (60% kcal from
DIO ly), 5TGM1 (1x10^6, intrave- linkage in obesity-induced tumor- [125]
fat), 15 weeks
nous) igenesis in myeloma
Oral carcinomas
HFD is able to boost the metastatic
HFD (60% kcal from T (Detroit-562 cells, orthotop- potential of CD36+ metastasis-
HFD [88]
fat), 7 days ic,) initiating cells to promote cancer
metastasis
OPEN ACCESS | www.cell-stress.com 58 Cell Stress | MAY 2021 | Vol. 5 No. 5X.-Z. Liu et al. (2021) Obesity and cancer
TABLE 1 (continued). Overview of in vitro and in vivo studies.
Obese tumor phenotype/ pro-
Obesity model Diet, duration Cancer model Ref
posed mechanism (keywords)
Ovarian cancer
T (coinjection: SKOV3ip1 cells + Adipocytes promote ovarian cancer
- - human adipocytes, subcutane- metastasis and provide energy for [89]
ous) rapid tumor growth
Pancreas cancer
Inflammation (TNFa signalling) and
HFD (60% kcal from GEMM (KC, 48Cre-K-rasLSL- increased fatty acid mitochondrial
DIO [99]
fat) G12D/+) beta-oxidation links obesity to tu-
mor promotion
Altered adipokine milieu and insu-
ob/ob, db/db - T (Pan02, 2,5*10^5, sub.c) lin resistance promotes cancer [126]
growth and dissemination
Activation of Kras via COX2 leads
HFD (60% kcal from GEMM (KC, KrasG12D, LSL-
to pancreatic inflammation and
DIO fat), >30 days/until Kras/Ela-CreERT and LSL- [98]
fibrosis and developement of
endpoint Kras/PDX1-Cre mice)
PanINs and PDAC
Increase in inflammatory cells,
HFCD (40% kcal from GEMM (KC, PDX1-Cre;LSL- cytokines, chemokines, and stro-
DIO [127]
fat), 3 months KRASG12D) mal fibrosis accelerates early pan-
creatic neoplasia
PIGF/VEGFR-1 signaling promotes
HFD (60% kcal from T (Pan02, AK4.4, graft, ortho- macrophage polarization and ac-
DIO [97]
fat), 10 weeks topic) celerated tumor progression in
obesity
Obesity-induced inflammation and
HFD (60% kcal from T (Pan02, AK4.4, graft, ortho- desmoplasia promote pancreatic
DIO, ob/ob [42]
fat), 10 weeks topic) cancer progression and resistance
to chemotherapy
Depletion LCN2 diminishes ECM
HFD (60% kcal from
GEMM (KC, KrasG12D/Cre) and deposition, immune cell infiltraton,
DIO fat), >50 days/until [128]
T (cells from KPC model) PanIN formation, and tumor
endpoint
growth
Mitochondrial arginase (ARG2) is
T (xenograft: 10^5 to 10^6 cells,
HFD (60% kcal from induced upon obesity and scilenc-
DIO syngeneic (KPC): 2,5-5*10^4, [94]
fat), 8 weeks ing or loss suppresses tumorigene-
orthotopic), GEMM (KPC)
sis
HFCD increase cancer incidence,
HFCD (40% kcal from GEMM (KC,P48+/Cre;LSL- fibrosis and inflammation of KC
DIO [129]
fat), >3 months KRASG12D) mice in addition to reducing au-
tophagic flux of PanIN lesions.
Obesity suppresses cell-
HFD (60% kcal from competition-mediated apical elimi-
DIO NA (CK19-RasV12-GFP) [130]
fat), 3 months nation of RasV12-transformed cells
from epithelial tissues
HFD heightens aerobic glycolysis
HFD (61,6% kcal
DIO GEMM (KC, KRASG12D/+) through hyperactivation of onco- [93]
from fat), 10 weeks
genic KRAS
GEMM (KC, fElasCre- KRAS reduces expression of FGF21
HFD (60% kcal from
DIO ERT;KrasLSL-G12D/+, in acinar cells to promote tumor- [131]
fat), 10 weeks
Ptf1aCreERT;KrasLSL-G12D/+) igenesis in mice on HFD
Endocrine-exocrine signaling drives
Ob/ob, db/db - GEMM (KC crossed with Ob/ob) [7]
obesity-associated PDAC
OPEN ACCESS | www.cell-stress.com 59 Cell Stress | MAY 2021 | Vol. 5 No. 5X.-Z. Liu et al. (2021) Obesity and cancer
TABLE 1 (continued). Overview of in vitro and in vivo studies.
Obese tumor phenotype/ pro-
Obesity model Diet, duration Cancer model Ref
posed mechanism (keywords)
Prostate cancer
HFD promotes prostatic basal-to-
luminal differentiation and acceler-
HFD (42% kcal from
DIO - ates intiation of protstate epithelial [132]
fat), 4 months
hyperplasia originated from basal
cells
High-fat diet fuels prostate cancer
HFD (60% kcal from
GEMM (FVB-Tg(ARR2/Pbsn- progression by rewiring the
DIO fat), from 3 weeks [117]
MYC)7Key/Nci) metabolome and amplifying the
until endpoint
MYC program
CICM – chemical induced cancer model; DIO – diet-induced obesity; GEMM – Genetically engineered mouse model; HFD – high fat diet; T –
transplant model; unless otherwise listed, duration of feeding indicates feeding pattern prior to transplantation or induction of cancer (e.g.
administration of agents to induce expression of tumor promoters or exposure to carcinogens).
for ten to twelve weeks prior to cancer challenge (Table 1). To establish HFD-induced obese mouse models, the
The long-term establishment period typically mimics the feeding period varies from four to 24 weeks, although ten
gradual development of obesity in humans as well as obe- to twelve weeks is commonly used (Table 1). This period is
sity-induced comorbidities that follows human obesity. On sufficient to induce obese phenotypes featuring increased
the other hand, this model is confounded by the potential body weight, elevated fasting glucose levels, hyperinsu-
biases created by the chosen dietary composition [9]. This linemia, insulin resistance, hepatic steatosis, dyslipidemia
could affect tumor outcomes independently of the obese and hypertension [19]. Such long-term HFD feeding is fairly
phenotype per se and has led many investigators to include consistently associated with enhanced tumor initiation and
both DIO and genetic models [10-13]. While findings from growth. In contrast, shorter HFD feeding periods of just
the two independent model systems generally overlap, four days is not associated with changes in body weight or
opposing results have been reported for intestinal cancer. features of metabolic syndrome. Such short-term feeding
In this study the authors reported that DIO induced intesti- regiments do not affect tumor initiation of orthotopically
nal tumor stemness, the db/db system led to a reduction in implanted mouse breast cancer cells [20]. This was evident
intestinal stem cell counts [12]. even though the HFD exposure was maintained for seven
Given that the majority of comorbidities seen in obese weeks until the end of the experiment [20]. Thus, an estab-
individuals are dependent on obesity-induced inflamma- lished state of obesity and obesity-induced physiological
tion [14], the field has relied on syngeneic transplants ra- changes are required for affecting tumor outcomes. Short
ther than xenograft models. Major challenges associated feeding periods should thus be avoided [20, 21], and the
with syngeneic models include limited tumor heterogenei- establishment of obesity-induced clinical and biochemical
ty, variable phenotypes depending on injection site, and features should be verified before tumor challenge.
relatively few transplantable cell lines as exemplified by an
extreme reliance on E0771, a murine mammary carcinoma Reversibility of obesity-induced effects on tumor out-
cell line and originally isolated as a spontaneous tumor comes
from C57BL/6 mouse [15] (Table 1). Finally, the clinical A key question in the field is to what degree reversing the
specificities in the links between obesity and cancer create obese phenotype affects tumor outcomes. In the ob/ob
additional challenges in the model setup. Perhaps the best model, reestablishing leptin expression through adenoviral
example of such is the dichotomy between obesity-induced expression in muscle reversed the obese phenotype and
breast cancer in pre- and postmenopausal women. Ovari- related biochemical parameters, leading to the suppression
ectomy is a well-established method to induce post- of pancreatic cancer progression [7]. In the same study, the
menopausal state in mice [16]. After removal of the ovary, enhanced tumor progression in the ob/ob model was also
endogenous estrogen production stops and the ovariecto- reversed by dietary caloric restriction. With the DIO models,
mized mice usually develop increased body weight, hepatic a number of studies have utilized dietary switch experi-
steatosis, insulin resistance and impaired glucose tolerance ments. Five weeks low-fat diet (LFD) feeding after estab-
[17]. Subcutaneously implanted tumors grow faster in the lished DIO was enough to reverse obesity-induced inflam-
ovariectomized model, regardless of diet, and this makes it mation and breast tumor formation [20]. A comparison
difficult to distinguish the effects of high-fat diet (HFD) on between a short-term and a long-term dietary switch
tumor progression from the effects of postmenopausal loss demonstrated that exposing the DIO mice to LFD for seven
of hormones [18]. days was not sufficient to affect the obesity-induced ef-
OPEN ACCESS | www.cell-stress.com 60 Cell Stress | MAY 2021 | Vol. 5 No. 5X.-Z. Liu et al. (2021) Obesity and cancer
fects on intestinal stem cells, while the phenotype was as metabolic reprogramming [33] and reactive oxygen spe-
reversed after four weeks LFD [12]. Taken together, these cies production [34, 35].
observations indicate that obesity-induced effects on tu- In females, higher leptin concentrations are associated
mor outcomes are reversible. with increased risk, as well as grade, stage and recurrence
of breast cancer [36, 37]. In the obese context, leptin has
MOLECULAR MECHANISMS OF THE OBESITY-CANCER been linked to tumor-initiating cell survival and obesity-
CONNECTION associated triple negative breast cancer development by
The interaction between obesity and cancer could be gov- promoting cancer stem cell enrichment and epithelial-to-
erned by genetic as well as non-genetic alterations. Cur- mesenchymal transition (EMT) [38, 39]. Leptin governs the
rently there is limited evidence for a genetic component stem cell phenotype through epigenetic mechanisms con-
linking obesity to cancer initiation. This likely reflects the trolled by a leptin-STAT3-G9a histone methyltransferase
difficulty of obtaining body mass index (BMI) measures as signaling axis [40].
well as incorporation of potential cachexia-induced weight In sum, several studies support a role of leptin in tu-
loss in large consortia databanks containing patient ge- morigenesis which favors cell growth and survival by in-
nomic status. Instead, an expanding number of cellular creasing proliferation and decreasing apoptosis, regulating
processes have been suggested to govern obesity effects in inflammatory processes, modulating cancer stem cell
malignancies. properties as well as metabolic activity. However, aug-
mented tumor formation is observed for several cancers,
Adipokines affecting carcinogenesis including cancers of the liver, kidney and pancreas in ob/ob
Adipose tissue is an active endocrine organ which secretes and db/db compared with wildtype mice [7, 11, 41, 42]
adipokines, chemokines and constituents of the extracellu- suggesting leptin-independent mechanisms. A number of
lar matrix (ECM) [22]. In response to increased demand of the early mammary cancer studies were performed with
triglyceride storage, adipose tissue undergoes hyperplasia the mouse mammary tumor virus (MMTV)- transforming
and hypertrophy in the transition from a lean to an obese growth factor-alpha (TGF-α) transgenic mouse model - a
state. These architectural adjustments induce changes in cancer model in which TGF-α overexpression is under the
the adipocyte secretome and released metabolites which control of the MMTV promoter. Without any further ge-
contribute to an altered micro- and macroenvironment in netic alteration, 30-40% of MMTV-TGF-α mice develop
the obese compared to the lean setting [22]. spontaneous tumors within 16 months [43]. However, the
overexpression of TGF-α in the background of either ob/ob
Leptin or db/db failed to develop tumors, suggesting a required
Adipokines have long been hypothesized to provide a role of leptin signaling in TGF-α driven carcinogenesis [44,
mechanistic link between cancer and obesity. Leptin is se- 45]. Interestingly, MMTV-Wnt1 cell-derived syngeneic
creted in proportion to adipose volume and total fat mass breast cancer cells displayed increased mammary tumor
[23, 24]. By signaling through leptin receptors in the central burden upon implantation into db/db mice, whereas im-
nervous system (CNS), leptin regulates energy balance by plantation of the same cells into ob/ob mice did not affect
functioning as a satiety signal, which reduces food intake tumor growth [38]. This suggests that peripheral cancer
and increases energy expenditure. In obese individuals, cell leptin signaling is required for obesity-dependent ef-
however, the satiety-promoting effects of leptin are im- fects of MMTV-Wnt1 cells. Taken together, these experi-
paired by the induction of cellular leptin resistance [25]. ments illustrate that leptin does not solely account for
Along with decreasing energy expenditure and promoting obesity-accelerated tumor progression for all cancer types.
obesity, hyperleptinemia has pronounced peripheral ef-
fects on cancer cells (discussed below) and the tumor mi- Adiponectin
croenvironment including immune cells – particularly the In addition to leptin, deregulation of the adipokine adi-
T-helper 1 cells [26, 27]. ponectin has been implicated in driving tumor progression
Leptin and the leptin receptors have been identified in in several obesity-associated cancer types, including colon,
malignant cells of various origins including hepatocellular liver, breast, renal, gastric, esophageal, pancreatic and
cancer, colorectal cancer, thyroid cancer and breast cancer endometrial cancer [46]. Circulating adiponectin levels
[28]. Overall, leptin has been assigned a pro-tumorigenic negatively correlate with body fat mass and adiposity. This
function. In breast cancer, leptin and the leptin receptor hormone acts as an insulin sensitizer of tissues such as liver
are overexpressed in primary and metastatic lesions com- and muscle, as well as balancing glucose and lipid metabo-
pared to non-cancer tissues [29, 30]. In cell culture, leptin lism [47, 48]. Adiponectin mediates its effects through its
acts as a growth-stimulating agent for breast cancer cells, classical and ubiquitously expressed receptors AdipoR1 and
promoting proliferation and repressing apoptotic pathways AdipoR2, and the non-classical receptor T-Cadherin [49, 50].
[31]. A similar role has been reported in colon cancer, Several signaling pathways are stimulated by adiponectin,
where leptin acts as a growth factor at stages correlating including AMPK, PI3K/AKT, MAPK, mTOR, NF-kB and STAT3
with tumor initiation in a murine model [32]. Moreover, [46]. Adiponectin exerts both anti-inflammatory and anti-
leptin treatment of cancer cells modulates processes such proliferative effects and is hence often referred to as the
“guardian angel adipokine” [48, 51]. In addition, adiponec-
tin also potently stimulates ceramidase activity through
OPEN ACCESS | www.cell-stress.com 61 Cell Stress | MAY 2021 | Vol. 5 No. 5X.-Z. Liu et al. (2021) Obesity and cancer
AdipoR1 and AdipoR2, and thereby enhance pro-apoptotic mouse models and human patients. A high intratumor
ceramide catabolism leading to formation of its down- E1:E2 ratio cooperates with NFκB pathway to increase tu-
stream anti-apoptotic metabolite sphingosine-1-phosphate mour stemness in obesity [65].
(S1P) [52]. HFD is sufficient to induce expression of the In summary, accumulating evidence obtained by a vari-
enzyme that generates S1P (sphingosine kinase 1, SphK1) ation of model systems supports an important role of adi-
and its receptor S1P receptor 1 (S1PR1) in syngeneic and pokines in tumor progression. On the other hand,
spontaneous breast tumors [53]. Targeting the knowledge derived from a curious counterpart to the
SphK1/S1P/S1PR1 axis in these models attenuates obesity- obese phenotype, the fatless A-Zip/F1 mouse, challenges a
induced tumor progression [53]. universal role. The fatless mouse model mimics generalized
Epidemiological studies have linked low serum adi- lipodystrophy and is characterized by the majority of the
ponectin levels to an increased risk of colon cancer [54]. comorbidities associated with obesity including insulin
This is consistent with in vivo studies that have demon- resistance and ectopic lipid accumulation but lack the se-
strated promotion of intestinal carcinogenesis by lack of cretion of adipokines. In chemical carcinogenesis and
adiponectin in both genetic and chemically induced cancer transgenic C3(1)/T-Ag mammary models, tumor incidence
models [55]. Another study also found a repressing role on was consistently higher in the fatless mouse compared
colonic epithelial proliferation in a chemically induced can- with controls - suggesting that adipocyte-independent fac-
cer model. However, this effect was specific to adiponec- tors drive tumorigenesis in this environment [66]. Overall,
tin-deficient mice fed an HFD. No effect was observed for more model-specific research is needed to fully understand
adiponectin-deficient mice fed a basal diet compared with the link between obesity, cancer and adipokines.
wild-type mice [56]. Likewise, adiponectin protects against
liver tumorigenesis in nude mice, and its reduced expres- Insulin and insulin-like growth factor-1 (IGF-1)
sion is associated with poor prognosis in obese patients Increased serum concentrations of insulin and IGF-1 are
with hepatocellular carcinoma [57]. These findings were frequently detected in obese individuals. Both factors have
supported by studies using an orthotopic liver tumor nude been proposed to serve as key hormonal mediators mech-
mouse model, which showed inhibited tumor growth and anistically linking obesity and cancer [67]. Insulin and IGF-1
lower incidence of lung metastasis in adiponectin treated are closely related ligands that activate the insulin receptor
mice [58]. Furthermore, adiponectin treatment suppressed (INSR) and IGF-1 receptor (IGF-1R), leading to stimulation
hepatic stellate cell activation and macrophage infiltration of two key cell proliferation and protein synthesis path-
[58]. In addition to anti-inflammatory and growth- ways for tumorigenesis - the PI3K-AKT-mTOR signaling
suppressing effects, adiponectin has been shown to con- pathway and the RAS-MAPK pathway (reviewed in [68]).
strain tumor growth by inhibiting tumor vasculature [58, Expression of the respective receptors is detected on can-
59]. On the contrary, lack of adiponectin in a MMTV- cer cells of different origins and several cancers are driven
polyoma middle T antigen (PyMT) model significantly re- by insulin and IGF-1 in vitro [69-73]. A positive association
duced tumor growth and angiogenesis [60, 61]. Thus, a is also detected between insulin and obesity-prone cancers
general role of adiponectin in tumor angiogenesis remains in terms of tumor growth in rodents as well as stage at
to be defined. diagnosis and death in humans (reviewed in [74]). Moreo-
ver, case-control and cohort studies have demonstrated
Other adipokines that individuals with higher levels of insulin or C-peptide
In addition to altered levels of leptin and adiponectin, in- (indirect insulin measure) are at higher risk for developing
creased levels of obesity-related adipokines such as inter- obesity-related cancers including breast, endometrial, col-
leukin-6 (IL-6), interleukin-8 (IL-8), tumor necrosis factor-α orectal, pancreatic, liver, ovarian and gastric cancers com-
(TNF-α), visfatin and resistin are associated with increased pared to individuals with low levels of these factors (re-
cancer risk [62]. Resistin is mainly expressed in adipocytes viewed in [75]).
and immune cells [63] and has been reported to promote In addition to a mitogenic function in carcinogenesis,
breast tumorigenesis in vitro and in vivo [64]. In a DIO insulin has been suggested to effect metabolic processes in
mouse model, its expression and secretion are significantly cancer cells. Insulin increases mitochondrial glucose oxida-
increased in mammary adipose tissue. Mechanistically, tion and augments cell division in cells derived from obesi-
HFD-induced elevated levels of circulating FFA promote ty-associated tumors, including colon, breast and prostate
peroxisome proliferator-activated receptor γ (PPARγ) sig- cancer. In contrast, no alteration of substrate preference is
nalling, which is an upstream regulator of WW-domain- observed in obesity-independent cell lines (melanoma,
containing transcription regulator 1 (WWTR1; also known lymphoma and small cell lung cancer) [76]. In studies using
as TAZ) expression. The enhanced TAZ expression via the an insulin lowering agent, dapagliflozin, a sodium-glucose
FFA/PPARγ axis further activates resistin secretion [64]. cotransporter-2 (SGLT2) inhibitor, reduction in insulin lev-
Adipose tissue is a major component of the breast. The els slowed obesity-accelerated tumor growth of both
cytokine secretion from adipocytes is affected by meno- syngeneic breast and colon cancer models. The authors
pausal status, which could link to the paradoxical associa- concluded that this effect is not due to increases in ketosis
tion of obesity and pre- and post-menopausal breast can- or to a direct effect on tumor cell division, but rather is
cer risk. After menopause, a high estrone (E1): estradiol mediated by the reversal of hyperinsulinemia, resulting in
(E2) ratio in tissue and circulation was observed in both diminished tumor glucose uptake and oxidation [77]. At
OPEN ACCESS | www.cell-stress.com 62 Cell Stress | MAY 2021 | Vol. 5 No. 5X.-Z. Liu et al. (2021) Obesity and cancer
present, several human clinical trials are investigating insu- ative breast cancer cell adhesion, migration and invasion
lin-lowering treatments as adjuvants to cancer treatments [83].
(reviewed in [74]). Furthermore, obesity-associated ECM remodeling has
been shown to regulate the properties of immune cells.
ECM remodeling and fibrosis Culturing of bone-marrow-derived macrophages on ECM
Tumors are composed of cancer cells along with their con- isolated from obese mice induce proliferation, changes the
nected stromal compartment. The tumor stroma consists morphology, impact polarization as well as promote angio-
of vasculature, non-transformed cell types including fibro- genic traits compared to cells cultured on top of ECM de-
blasts and immune cells, as well as structural elements rived from lean mice [85]. Hermano et al. reported that
such as the basement membrane and ECM. The stromal macrophage function could be regulated by heparanase,
compartment is an integral part of cancer initiation, an endoglucuronidase that cleaves heparin sulfate in ECM
growth and progression (reviewed in [78]) and a number of [86]. They provided evidence that heparanase preferently
studies have begun to delineate how obesity-induced is expressed in clinical/experimental obesity-associated
stromal alterations influence tumor growth. Adipose tissue breast tumors, and that heparanase deficiency abolishes
in obese individuals is among others characterized by an obesity-accelerated orthotopic tumor growth. In the obese
altered biochemical as well as biophysical microenviron- environment, they showed that heparanase stimulates
ment [79]. For instance, adipocytes in expanding adipose macrophage production of inflammatory mediators that
tissue deposit altered amounts of ECM components caus- induce aromatase, a rate-limiting enzyme in estrogen syn-
ing ECM remodeling and changes in tissue stiffness. Fur- thesis [86].
thermore, obese tissue ECM contains more aligned fibers
and higher interstitial pressure (reviewed in [80]). These Cancer metabolism
changes are not restricted to subcutaneous and visceral fat Metabolic reprogramming is commonly observed in cancer
but occur also in other fat depots. For instance, the home- cells during tumor progression. Given the multiple local
ostasis of mammary fat is disrupted in obese mouse mod- and systemic metabolic abnormalities in obese individuals,
els, as this tissue is enriched in myofibroblasts and stiff- the obese state provides an interesting case for new dis-
ness-promoting ECM components [81]. These alterations, coveries in the interface between systemic and tumor me-
which are characteristic features of desmoplasia and fibro- tabolism.
sis, can promote the tumorigenic potential of premalignant
human breast epithelial cells [81]. Research on mouse The effects of FFA on tumor initiation
models has shown that obesity also promotes desmoplasia Recent studies have proposed that lipid metabolism and
in the pancreas, a common histological feature of PDAC, fatty acid oxidation contribute to cancer stemness. A sub-
which is associated with accelerated tumor growth and population of human carcinoma cells that expressed high
impaired delivery/efficacy of chemotherapeutics through levels of the fatty acid transporter CD36 and lipid metabo-
reduced perfusion [42]. Interestingly, it was suggested that lism genes was identified to possess cancer stemness fea-
the exacerbated desmoplasia and augmented tumor tures and to contribute to a poor prognosis in human can-
growth was a result of crosstalk between adipocytes, tu- cer patients [87, 88]. In intestinal cancers, exposure to FFA
mor- associated neutrophils and pancreatic stellate cells was demonstrated to activate PPARδ-dependent signaling
[42]. Grohmann et al. reported how obesity-associated to endow tumor-initiating capacity [12]. In addition to di-
hepatic stress could independently contribute to the path- rect cancer cell-autonomous effects, obesity-induced ele-
ogenesis of non-alcoholic steatohepatitis (NASH), fibrosis vated levels of saturated fatty acid drive the accumulation
and hepatocellular carcinoma (HCC). Obesity promotes of metabolically activated macrophages in adipose tissue.
hepatic STAT1 dependent T-cell infiltration, NASH and fi- Such macrophages were shown to enhance stem-like
brosis as well as NASH-independent STAT3-dependent HCC properties in triple negative breast cancer cells and pro-
[82]. mote tumor initiation in obese mouse models [20]. Overall,
Moreover, single ECM components that are deregulat- there is compelling evidence that fatty acids could be a
ed in the obese state have been demonstrated to promote critical link between obesity and tumor initiation.
tumorigenesis. One example is collagen VI, which is abun-
dantly produced and secreted by adipocytes. A recent The effects of FFA on tumor progression
study applying proteomic analysis of ECM isolated by in In addition to enhanced tumor initiation, emerging evi-
vitro decellularization methods identified collagen VI to be dence indicates that the elevated circulating FFA and fatty
up-regulated in murine obese mammary gland and breast acid binding proteins in the obese state are associated with
tumor tissues relative to lean tissues [83]. Adipocyte- cancer progression. In ovarian cancer, the rapid growth
derived collagen VI has previously been demonstrated to and metastatic colonization of cancer cells were suggested
be important for early mammary tumor progression, as to be directly fueled by fatty acids delivered by fatty acid
collagen knockout mice in the background of the MMTV- binding protein 4 (FABP4), also known as adipocyte FABP
PyMT mammary cancer model have reduced rates of early (A-FABP), from adipocytes [89]. Furthermore, lipolysis in
hyperplasia and primary tumor growth [84]. In vitro studies adipocytes and increased β-oxidation rate in cancer cells
point to that full-length collagen VI is a driver of triple neg- were observed in co-culture experiments, suggesting that
adipocyte-derived fatty acids can act as an energy source
OPEN ACCESS | www.cell-stress.com 63 Cell Stress | MAY 2021 | Vol. 5 No. 5X.-Z. Liu et al. (2021) Obesity and cancer
fueling cancer cells [89]. Hao et al. expanded the role of Inflammation
FABP itself. They demonstrated that increased circulating Obesity is associated with local and systemic immune sys-
levels of A-FABP enhanced breast cancer stemness and tem dysregulation characterized by increased secretion of
aggressiveness in both in vitro and in vivo models [90]. inflammatory cytokines and phenotypic conversions in
Moreover, the authors further determined that the associ- immune cells [96]. Recently, a growing number of studies
ation is dependent on STAT3/ALDH1 signaling regardless of have suggested that obesity-associated inflammatory al-
endogenous A-FABP level, suggesting a dual mechanism by terations might promote progression of multiple cancer
which A-FABP supports tumor progression both as a fatty types through diverse mechanisms.
acid transporter and as a signaling molecule [90]. Consist-
ently, Madak-Erdogan et al. demonstrated that obesity- Tumor immune cell abundances
induced elevated circulating FFA could be taken up by es- A fundamental question is whether obesity-induced chron-
trogen-positive breast cancer cells and promote cancer ic inflammation alters immune cell infiltration in tumors. At
proliferation in obese postmenopausal women [91]. Also, the histological level, HFD-feeding increases infiltration of
these findings suggested that fatty acids activate a mTOR tumor-associated macrophages in both transplant and
and MAPK dependent signaling network to facilitate in- some autochthonous PDAC models [97-99] as well as in
creased glycolytic and aerobic respiration in breast cancer transplant models of breast cancer [53, 86] and in chemi-
cells. Finally, cancer cells and immune cells display distinct cally induced hepatocarcinoma [100, 101]. In contrast,
metabolic adaptations in the obesogenic tumor microenvi- transcriptional analysis of the duodenum of K-rasG12Dint
ronment [92]. For example, HFD represses PHD3 expres- mice revealed down regulated F4/80 expression in mice
sion specifically in cancer cells, which rewire their metabo- fed HFD [102], and fluorescence- activated cell sorting
lism to accelerate fatty acid uptake and oxidation. This in (FACS) analysis of transplanted E0771 breast tumors
turn impacts the fatty acid availability for T cells in the showed a reduction of CD11b+F4/80+ macrophages [103].
same microenvironment and thereby impairs T cells infil- In kidney cancer, increased numbers of inhibitory dendritic
tration and function [92]. cells (DC) infiltrate tumors of DIO mice [104]. Similar ob-
servations have been reported in breast cancer, as a popu-
Other metabolites and tumor progression lation of CD11b+F4/80− cells (consisting of neutrophils and
In addition to fatty acid-related adaptations, modifications monocytes) identified by FACS showed a 31% increase in
in other metabolic pathways have been identified in tu- E0771 tumors from HFD mice [103]. Taken together, con-
mors evolving in obese environments, including glucose flicting results on obesity-induced immune cell tumor infil-
handling, nitrogen metabolism and pyruvate-dependent tration has been reported for different cancer types as well
mitochondrial respiration. For example, in a genetic model as for different cancer/obesity models applied. This is in
for pancreas cancer, HFD feeding heightened aerobic gly- line with our own preliminary findings. We interrogated
colysis through hyperactivation of oncogenic KRAS [93]. the immune-infiltrating cells in tumors grown in obese and
Moreover, transcriptomic analysis of an orthotopic xeno- non-obese mouse models using a 36-marker immune-
graft PDAC model revealed an enrichment of nitrogen me- focused mass cytometry panel [105]. Immunotyping of
tabolism pathways [94]. The authors identified the mito- three syngeneic breast cancer models and two pancreas
chondrial enzyme arginase 2 (ARG2), which catabolizes cancer models revealed that tumor immune infiltrate com-
arginine into ornithine and urea, to be induced in obese position is highly model- and cancer type-specific. While no
mouse tumors and that its expression level correlated with major immune cell alterations were observed in the pan-
patient BMI. As an important enzyme at the final step of creas cancer models and in two of the breast cancer mod-
the urea cycle, the obesity-induced upregulated ARG2 en- els, HFD-feeding increased two T cell suppressive cell types
hances nitrogen flux into the urea cycle. Moreover, the and decreased CD8 T-cells in the E0771 breast cancer
depletion of ARG2 causes ammonia accumulation and sup- model [105].
presses PDAC, particularly in obese hosts [94]. In a search
for mechanisms underlying the increased cancer risk that is Macrophages
associated with the combination of metabolic deregulation In addition to affecting the overall infiltration of immune
and circadian disruption, Ramos et al. demonstrated that a cells, accumulating evidence indicates that obesity affects
non-canonical function of BMAL1 limits obesity-promoted immune function through modulating immune cell pheno-
triple-negative breast cancer [95]. BMAL1, a key circadian types. The immune subset that has so far received the
transcription factor, suppresses the flexibility of mitochon- most attention is macrophages. In several cancer models,
drial substrate usage and pyruvate-dependent mitochon- obesity has been proposed to regulate recruitment, polari-
drial respiration induced by chronic insulin treatment in zation and signaling of macrophages, thereby contributing
vitro. Interestingly, orthotopic transplantation of E0771 to accelerated tumor progression. In breast and pancreatic
breast cancer cells depleted for BMAL1 revealed that cancer models, obesity induces recruitment of tumor-
BMAL1 functions as a tumor suppressor in obese, but not associated macrophage (TAM) with an M2-like cytokine
in lean mice. In humans, down-regulation of BMAL1 is as- profile. VEGFR-1 is abundantly expressed in TAMs, and
sociated with higher risk of metastasis [95]. blockade of VEGFR-1 signaling in obese but not lean mice
leads to a shift in pro-tumor cytokine production and TAM
polarization from an M2 pro-tumor to an M1 anti-tumor
OPEN ACCESS | www.cell-stress.com 64 Cell Stress | MAY 2021 | Vol. 5 No. 5X.-Z. Liu et al. (2021) Obesity and cancer
phenotype that ultimately reduce obesity-induced tumor ways, through genetic depletion of key cytokines or phar-
progression [97]. Furthermore, the elevated level of fatty macological inhibition of specific immune cell populations
acids during obesity drives polarization of adipose tissue in animal models. Systemic knockout of TNF-α, IL-6 or
macrophages towards a metabolically activated phenotype. TNFR1 all prevent obesity-induced cancer formation in HCC
This phenotypic shift alters the niche to support triple neg- [100] and PDAC [99] suggesting a functional role of the
ative breast cancer stemness and tumorigenesis through immune apparatus. Although these studies are limited by
an IL-6/GP130 signaling axis [20]. In colitis-associated colo- the systemic depletion, they indicate a required role of the
rectal cancer, signaling through obesity-induced IL-6 was immune system in obesity-induced cancers.
reported to shift macrophage polarization towards a tu- Experimental evidence across cancer types supports a
mor-promoting phenotype that produced the chemokine pivotal role of cyclooxygenase-2 (COX-2)-mediated in-
CC-chemokine-ligand-20 (CCL-20) in the tumor microenvi- flammatory pathways in obesity promoted cancer progres-
ronment. CCL-20 in turn, promoted tumorigenesis by re- sion [98, 111]. A study applying an adult acinar-specific
cruiting CC-chemokine-receptor-6 (CCR-6)-expressing γδ mutant Kras PDAC model, demonstrated that high fat con-
T and B cells via chemotaxis [106]. Obesity-accelerated sumption can activate and sustain Kras activity via COX-2
cancer growth is also associated with increased IL-6 levels induction and COX-2 mediated positive feedback loops,
and shift in M2/M1 macrophage ratio in a genetic mouse which in turn contribute to the development of PanINs and
model of prostate cancer. Interestingly, these effects could PDAC. Inhibition and genetic knockout of COX-2 prevented
specifically be blocked in HFD-fed mice by inhibition of the pancreatic inflammation and initiation of PDAC, suggesting
IL-6 receptor [107]. important roles for COX-2-dependent inflammatory pro-
In two syngeneic orthotopic transplant models, a cesses [98]. In contrast, treatment with aspirin, a non-
NLRC4 inflammasome/IL-1β signaling was demonstrated to steroidal anti-inflammatory agent (NSAID) that blocks the
provide a link between obesity and breast cancer progres- cyclooxygenase enzymes, did not impact tumor progres-
sion. The obese tumor microenvironment induced an in- sion, immune cell infiltration or fibrosis in an obese genetic
crease in the number of tumor-infiltrating macrophages model of PDAC [7]. Consistently, the growth advantage of
with an activated NLRC4 inflammasome, which further orthotopically implanted oncogenic Kras-driven PDAC cell
activated IL-1β. The NLRC4/IL-1β module was able to pro- lines in the obese environment was sustained in obese
mote angiogenesis via up-regulation of VEGFA, which in C57Bl6 mice lacking T-, B- and NK-cells (Rag2-/-::CD47-/-
turn to contributed tumor progression [108]. In a liver can- ::Il2rg-/-) [105].
cer model, HFD induced liver endoplasmic reticulum (ER)
stress boosted macrophages-mediated production of in- Microbiome
flammatory cytokines. This in turn activated TNF receptor 1 An integrated part of the obese phenotype is alterations in
(TNFR1)-IκB kinase β (IKKβ) signaling, which thereby con- the microbiota composition [112, 113]. As a natural part of
tributed to hepatocyte proliferation and formation of HCC the tumor microenvironment of gastrointestinal malignan-
[101]. The importance of macrophages in the connection cies, the altered microbiota can directly participate in the
between obesity and breast cancer has however been chal- regulation of gastrointestinal cancer progression. In addi-
lenged. Bousquenaud et al. reported that HFD tumors con- tion to such local effects, microbiota-mediated metabolites
tained reduced amount of M1 macrophages, and that de- display both systemic metabolic and inflammatory changes
pletion of macrophages by clodronate liposomes boosted [114, 115]. Gut microbiota are the major producers of
tumor growth in HFD mice. Hence, the authors concluded short-chain fatty acids (SCFAs), which can be the utilized as
that macrophages do not contribute to promote obesity an energy source and as key signaling molecules. Feeding
accelerated tumor progression [103]. mice with an HFD results in the decreased production of
multiple SCFAs including acetate, propionate and butyrate
T cells and were demonstrated to promote G12D mutant Kras-
Tumoral T cell function is of great interest, especially since driven intestinal carcinogenesis [102]. Interestingly, butyr-
the T cell is the critical mediator of immunotherapies, in- ate supplement through feeding successfully reversed the
cluding adoptive T cell therapy and immune checkpoint cancer progression effect of the high-fat feeding. Also,
therapy. Obese cancer patients have been reported to dis- fecal-transplantation from HFD-fed to normal diet fed Kras
play increased levels of dysfunctional and exhausted T cells mutant mice was sufficient to induce tumorigenesis [102].
[92, 109], suggesting that immunotherapy could be a These findings suggest that an HFD-induced microbiota
promising treatment option for obese cancer patients. shift synergizes with the Kras mutation during tumorigene-
Indeed, in both human patients and preclinical animal sis and that such effects could act independently of obesity.
models, obesity associates with better response to anti- In chemically-induced HCC, obesity-dependent alterations
PD1 and anti-CD8 immune checkpoint therapy [92, 109, of the gut microbiota promote tumorigenesis through de-
110]. oxycholic acid - a secondary bile acid [11, 111]. This me-
tabolite could cause DNA damage and provoke a senes-
Blocking obesity-induced inflammation cence-associated secretory phenotype (SASP) in hepatic
To address whether inflammatory processes are required stellate cells to promote HCC development. In addition to
for obesity-induced tumor initiation and progression, sev- bile acids, the altered gut microbiota provided lipoteichoic
eral studies have obstructed inflammatory signaling path- acid to the liver, and this compound synergistically pro-
OPEN ACCESS | www.cell-stress.com 65 Cell Stress | MAY 2021 | Vol. 5 No. 5X.-Z. Liu et al. (2021) Obesity and cancer
moted the SASP in hepatic stellate cells. Also, lipoteichoic been reported in prostate cancer [117], colorectal cancer
acid is able to induce the COX-2-mediated immunosup- [10] and breast cancer [118]. How the obese environment
pression in liver immune cells [111]. Altogether, the micro- induces such changes is currently unknown, but it is clear
biota is an important component when studying gastroin- that it could have profound effects on tumorigenesis. In-
testinal cancers in obesity. Due to the complex environ- terestingly, many metabolic intermediates including Ace-
ment, further studies are required to understand and es- tyl-CoA and S-adenosyl methionine act as epigenetic pre-
tablish a solid mechanistic basis of the relationships be- cursors and can directly and indirectly affect chromatin
tween the microbiome, obesity and gastrointestinal can- modifications and function [119]. Going forward, it will be
cers progression. interesting to investigate if obesity-dependent changes in
cellular metabolites are potential drivers of chromatin re-
SUMMARY AND FUTURE PERSPECTIVES modeling in tumors.
As an expanding number of reports aiming to identify mo- While the epidemiological link between obesity and
lecular connections between the obese state and tumors cancer risk is well proven, the link between obesity and
are released, it is clear that the complexity of the obese metastatic spread is less clear. This is reflected in far fewer
environment likely translates into integrated and complex mouse studies of metastasis in the obese setting. In one
molecular drivers. This necessitates the use of proper study, breast cancer lung metastatic colonization was en-
model systems that realistically recapitulates such envi- hanced in obese mice via GM-CSF- and IL-5 dependent
ronments. Whereas we have so far relied on mouse models, recruitment of neutrophils [13]. Further studies in breast
additional model systems as for example organoids grown cancer using transplant and genetic models suggest that
in obese and non-obese conditions could become valuable obesity-dependent changes in the sphingolipid profile is
alternatives to alleviate the limitations linked to mouse driving metastatic colonization in the obese setting [53].
studies described above. However, potential mechanisms of obesity linked meta-
So far, the most consistent molecular link between the static spread are currently understudied.
obesogenic environment and tumorigenesis revolves Finally, in addition to potentially revealing new exciting
around increased abundance of fatty acids, ECM remodel- mechanistic pathways, discoveries of dependencies of the
ing and immune cell differentiation. Fatty acids have been obesity-cancer link might lead to the development of novel
implicated in both tumor growth and tumor initiation targeted therapies for the benefit of growing group of
through cell intrinsic and extrinsic mechanisms. In the tu- obese cancer patients.
mor cells FFA have been suggested to act as an alternative
fuel source while FFA have also been shown to modulate ACKNOWLEDGMENTS
metabolically-activated macrophages and T-cells to create We thank members of our laboratory and Anders Molven
a pro-tumorigenic microenvironment. Direct effects of the for insightful discussions. NH was supported by a Trond
obese environment on T-cells further translate into a po- Mohn Foundation Starter grant, The Norwegian Research
tential added benefit of immune checkpoint therapy in the Council (#275250) and the Norwegian Cancer Society
obese setting. Through tumor promoting modulation of (#207028). We apologize to colleagues whose important
the stromal compartment of the microenvironment, obesi- work was not cited due to space limitations. Figure 1 was
ty leads to both biophysical changes driven by alterations created using Biorender.
in myofibroblast cell populations and biochemically
through altered collagen VI secretion and processing. CONFLICT OF INTEREST
So far, the vast majority of mechanistic studies have fo- The authors declare no competing interests.
cused on obesity-dependent alterations in adipocytes, im-
mune cells and their secreted molecules. However, recent COPYRIGHT
work urges us to take a broader look at additional cell © 2021 Liu et al. This is an open-access article released
types that could be affected by the obese state. This, for under the terms of the Creative Commons Attribution (CC
example, include the islet cells of the pancreas. In the BY) license, which allows the unrestricted use, distribution,
obese setting, these adapt by inducing Cck which in turn and reproduction in any medium, provided the original
acts locally on pancreatic acinar to cells to drive tumor- author and source are acknowledged.
igenesis [7].
Another area of increasing interest is the connection
between obesity and epigenetic deregulation. This connec- Please cite this article as: Xiao-Zheng Liu, Line Pedersen and Nils
tion was previously demonstrated in somatic tissues and Halberg (2021). Cellular mechanisms linking cancers to obesity.
has recently also been investigated in tumors [116]. Epige- Cell Stress 5(5): 55-72. doi: 10.15698/cst2021.05.248
netic rearrangements in cancers grown in obese mice, have
OPEN ACCESS | www.cell-stress.com 66 Cell Stress | MAY 2021 | Vol. 5 No. 5You can also read

















































