Asthmatic bronchial smooth muscle increases rhinovirus replication within the bronchial epithelium
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Article
Asthmatic bronchial smooth muscle increases
rhinovirus replication within the bronchial
epithelium
Graphical abstract Authors
Pauline Esteves, Benoit Allard,
Alexis Celle, ..., Pierre-Olivier Girodet,
Thomas Trian, Patrick Berger
Correspondence
thomas.trian@u-bordeaux.fr
In brief
Esteves et al. report that asthmatic
bronchial smooth muscle cells
downregulate the PKR antiviral pathway
within bronchial epithelium. CCL20
expression is increased in asthmatic
bronchial smooth muscle cells. CCL20
inhibits the PKR antiviral pathway and
increased rhinovirus replication in
bronchial epithelium.
Highlights
d BSM remodeling is an important feature of severe asthma
pathophysiology
d PKR antiviral pathway is downregulated in asthmatic
bronchial epithelium
d CCL20, secreted by asthmatic BSM cells, is responsible for
PKR downregulation
d Severe asthmatic BSM cells can play a role in rhinovirus
infection of the bronchial epithelium
Esteves et al., 2022, Cell Reports 38, 110571
March 29, 2022 ª 2022 The Author(s).
https://doi.org/10.1016/j.celrep.2022.110571 ll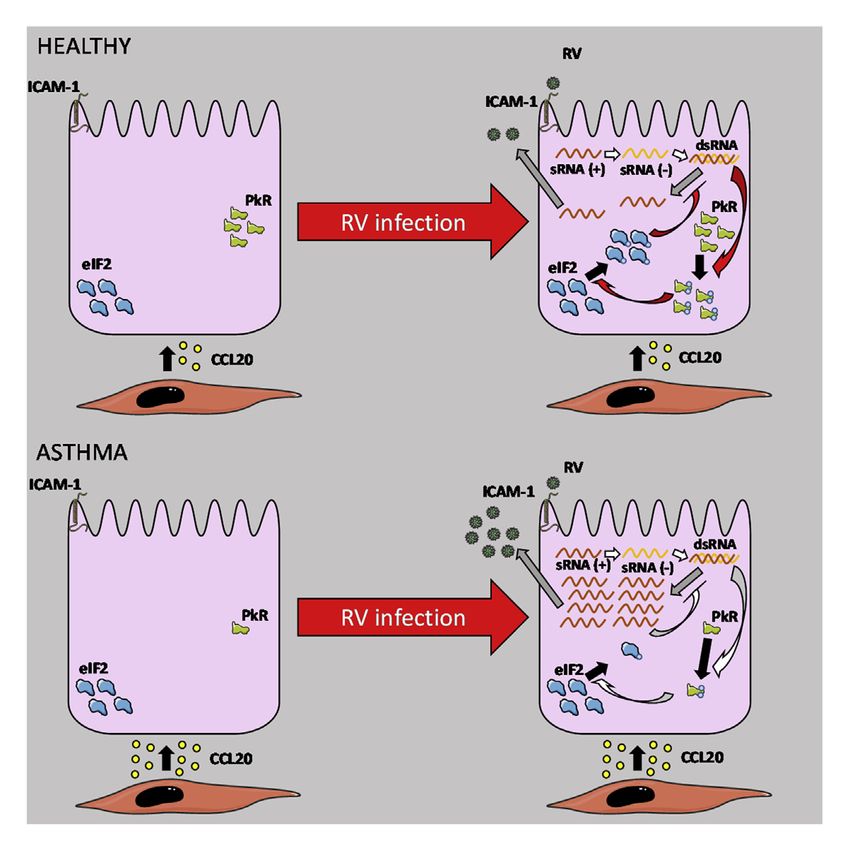
ll
OPEN ACCESS
Article
Asthmatic bronchial smooth
muscle increases rhinovirus
replication within the bronchial epithelium
Pauline Esteves,1,2 Benoit Allard,1,2 Alexis Celle,1,2 Isabelle Dupin,1,2 Elise Maurat,1,2 Olga Ousova,1,2
Matthieu Thumerel,1,2,3 Jean-William Dupuy,1,2 Thierry Leste-Lasserre,1,2 Roger Marthan,1,2,3 Pierre-Olivier Girodet,1,2,3
Thomas Trian,1,2,4,5,* and Patrick Berger1,2,3,4
1Univ-Bordeaux, Centre de Recherche Cardio-thoracique de Bordeaux, U1045, Département de Pharmacologie, CIC 1401, 33000 Bordeaux,
France
2INSERM, Centre de Recherche Cardio-thoracique de Bordeaux U1045, Plateforme Transcriptome Neurocentre Magendie U1215,
Functionnal Genomics Center (CGFB) Proteomics Facility, CIC 1401, PTIB - Ho ^ pital Xavier Arnozan, Avenue du Haut Léve
^que, 33600
PESSAC, 33000 Bordeaux, France
3CHU de Bordeaux, Service d’exploration fonctionnelle respiratoire, Service de pharmacologie, CIC 1401, Service de chirurgie thoracique,
33604 Pessac, France
4These authors contributed equally
5Lead contact
*Correspondence: thomas.trian@u-bordeaux.fr
https://doi.org/10.1016/j.celrep.2022.110571
SUMMARY
Rhinovirus (RV) infection of the bronchial epithelium is implicated in the vast majority of severe asthma exac-
erbations. Interestingly, the susceptibility of bronchial epithelium to RV infection is increased in persons with
asthma. Bronchial smooth muscle (BSM) remodeling is an important feature of severe asthma pathophysi-
ology, and its reduction using bronchial thermoplasty has been associated with a significant decrease in
the exacerbation rate. We hypothesized that asthmatic BSM can play a role in RV infection of the bronchial
epithelium. Using an original co-culture model between bronchial epithelium and BSM cells, we show that
asthmatic BSM cells increase RV replication in bronchial epithelium following RV infection. These findings
are related to the increased production of CCL20 by asthmatic BSM cells. Moreover, we demonstrate an orig-
inal downregulation of the activity of the epithelial protein kinase RNA-activated (PKR) antiviral pathway.
Finally, we identify a direct bottom-up effect of asthmatic BSM cells on bronchial epithelium susceptibility
to RV infection.
INTRODUCTION need in patients with severe asthma, whereas recent advances
in new biologic therapies have significantly decreased the exac-
Asthma is a very frequent airway disease that affects 6% to 20% erbation rate (Bel et al., 2014; Bleecker et al., 2016; Castro et al.,
of the population of Western European countries (To et al., 2012). 2018; Humbert et al., 2005; Ortega et al., 2014). Indeed, on the
Severe asthma, defined according to the last ATS/ERS (Amer- one hand, the use of these biologics is limited to a subgroup of
ican Thoracic Society/European Respiratory Society) task force, person with severe asthma (i.e., patients with allergies or high
affects 3% to 5% of all person with asthma but is responsible for levels of blood eosinophils or type 2 inflammation), whereas,
a large proportion of resource expenditure (Chung et al., 2014). on the other hand, a high proportion of patients on biologics
Histologically, bronchi from persons with severe asthma have continue to present some exacerbations, albeit at a lower rate
been characterized by various features of remodeling-associ- (Bel et al., 2014; Bleecker et al., 2016; Castro et al., 2018; Hum-
ated bronchial epithelial abnormalities, such as metaplasia, bert et al., 2005; Ortega et al., 2014).
thickened epithelium and reticular basement membrane, goblet The role of bronchial epithelium in asthma exacerbation has
cell hyperplasia or mucus hypersecretion (Gras et al., 2012), and been well documented. Indeed, viral infection of the bronchial
increased bronchial smooth muscle (BSM) mass (Trian et al., epithelium has been implicated in 80% of asthma exacerbations
2007). Functionally, BSM remodeling indicates a poor prognosis in children and adolescents and in 60% of those in adults (Busse
in asthma, since an increased BSM mass is associated with et al., 2010). In each case, the human rhinovirus (RV) was the
decreased lung function (Kaminska et al., 2009) and an dominant viral pathogen, and RV caused 60% of all virus-
increased exacerbation rate (Girodet et al., 2016). Clinically, induced asthma exacerbations (Busse et al., 2010). The suscep-
the reduction of asthma exacerbations remains a major unmet tibility of bronchial epithelium to RV infection appears to be
Cell Reports 38, 110571, March 29, 2022 ª 2022 The Author(s). 1
This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/).ll
OPEN ACCESS Article
Table 1. Clinical and functional characteristics of patients
Characteristics Non-asthma Asthma p value
No. of patients 40 20
Sex (M/F) 24/16 8/12 NS
Age (y) 61.5 ± 1.5 57.0 ± 2.3 NS
Range (y) 29–76 30–76
Body mass index (kg/m2) 22.6 ± 1.1 25.7 ± 1.2 NS
Treatments
LABA, no. of patients 0 20
ICS, no. of patients 0 20
OCS, no. of patients 0 3
FEV1
Liters 2.50 ± 0.14 1.86 ± 0.15 0.001
Percentage of predicted value 88.6% ± 4.14% 70.44% ± 5.22% 0.01
FVC
Liters 3.39 ± 0.15 2.66 ± 0.19 0.005
Percentage of predicted value 104.3% ± 4.13% 84.14% ± 4.94% 0.005
Plus or minus values are means ± SEM. LABA, long-acting beta-agonist; ICS, inhaled corticosteroid; OCS, oral corticosteroid; FEV1, forced expiratory
volume in 1 s; FVC, forced vital capacity; NS, non-significant. The p values were calculated with the use of a two-sided independent t test for variables
with a parametric distribution and the Mann-Whitney U test for comparison of nonparametric variables. Sex distribution p value was calculated using
chi-square test.
increased in persons with asthma (Zhu et al., 2019), but the with severe asthma were similar to non-asthmatic patients in
mechanism remains unclear even though intrinsic modifiers, terms of the sex ratio, age, and body mass index (BMI). Not sur-
such as upstream activators of interferon production, have prisingly, persons with asthma presented lower forced expira-
been demonstrated (Gielen et al., 2015; Girkin et al., 2015). tory volume in one second (FEV1) and forced vital capacity
Interestingly, BSM remodeling has shown a significant associ- (FVC) values, which were assessed as both the percentage of
ation with asthma exacerbations in both severe and non-severe the predicted value and the absolute value. Indeed, despite the
asthma. Bronchial thermoplasty, which has been developed to heterogeneity in the non-asthmatic group related to the smoking
reduce BSM mass, decreased the rate of asthma exacerbations status (active, former, or non-smoker), we paid special attention
after 3 months for up to 5 years in selected persons with severe to enroll a population of non-asthmatic patients with normal lung
asthma (Cox et al., 2007; Wechsler et al., 2013). Moreover, the function values for both FEV1 and FVC values and without any
addition of gallopamil for 12 months in the treatment of severe inhaled treatment.
asthma reduced both the BSM thickness and, subsequently,
the exacerbation rate during the 3-month follow-up compared Asthmatic BSM cells increased RV replication in
with placebo (Girodet et al., 2015). In addition, the rate of exac- bronchial epithelium
erbations was increased in persons with non-severe asthma with To assess the role of BSM cells in RV infections of bronchial
high BSM mass compared with that in patients with low BSM epithelial cells, we developed a co-culture model using differen-
mass after 1 year of follow-up (Girodet et al., 2016). However, tiated air-liquid interface bronchial epithelium with ciliated and
the mechanisms by which asthmatic BSM cells could play a mucus cells grown on top of BSM cells (Figure 1A). We applied
role in asthma exacerbations remain completely unknown. bronchial epithelial cells obtained from non-asthmatic (asth-
The goal of the present study was thus to assess the role of matic for Figure 1E only) on top of BSM cells obtained from either
asthmatic BSM in epithelial susceptibility to RV infection. Using non-asthmatic or asthmatic persons (Figure 1A). All experiments
both an in vitro co-culture model of bronchial epithelial cells were performed with one or two different non-asthmatic epithelia
and BSM cells and ex vivo patient biopsies, we showed that and each epithelium was only used in one set of experiments.
asthmatic BSM cells increased RV replication within the epithe- Then, after 2 days of co-culture of bronchial epithelial cells
lium. Moreover, we demonstrated that such an increase is medi- with BSM cells, we infected the bronchial epithelium with human
ated by the production of CCL20 by the BSM and involves the RV16 at a multiplicity of infection (MOI) of 0.1 for either 6 or 24 h
antiviral protein kinase RNA-activated (PKR) pathway. (Figure 1B). Moreover, this MOI emulated an in vivo infection of
the bronchial epithelium (Cakebread et al., 2011).
RESULTS After only 6 h of infection, the number of RV RNA particles de-
tected by digital PCR (Figure 1C) or RT-qPCR (Figure S1A) was
Patient characteristics not changed in bronchial epithelium co-cultured with asthmatic
The clinical and functional characteristics of both severe asthma BSM cells compared with that co-cultured with non-asthmatic
and non-asthmatic persons are presented in Table 1. Persons BSM cells. Such a short time interval allowed the RV to enter
2 Cell Reports 38, 110571, March 29, 2022ll
Article OPEN ACCESS
(legend on next page)
Cell Reports 38, 110571, March 29, 2022 3ll
OPEN ACCESS Article
the bronchial epithelial cells but did not allow its replication (Kim Asthmatic BSM cells downregulated PKR expression in
et al., 2015). This first result showed that asthmatic BSM cells did bronchial epithelium
not allow increased RV entry into bronchial epithelium, suggest- To identify the mechanisms of the BSM-dependent increase in
ing that its infectivity by RV was not modified by co-culture with RV RNA particles within the bronchial epithelium, we assessed
asthmatic BSM cells compared with that with non-asthmatic the interferon response after RV infection. Surprisingly, we did
BSM cells. Moreover, this first conclusion was reinforced by not find any modification of interferon-b production (Figure S4A)
the absence of change in the expression of ICAM1, which is or interferon-induced genes (i.e., GBP1, ISG15, and OAS1) in
the main route of entry of RV, in bronchial epithelium co-cultured bronchial epithelial cells co-cultured with asthmatic BSM cells
with BSM cells from either asthmatic or non-asthmatic persons compared with that in those co-cultured with non-asthmatic
at both the mRNA and protein levels (Figures S2A and S2B). BSM cells (Figures S4B–S4D).
In contrast, the number of RV RNA particles was largely Since PKR, also known as an interferon-induced double-
increased 24 h after infection compared with that observed after stranded RNA-dependent protein kinase, is an important
6 h in bronchial epithelium co-cultured with either asthmatic or antiviral protein kinase (Garcia et al., 2007), we assessed its
non-asthmatic BSM cells (Figures 1C and 1D). This result expression both in vitro and ex vivo. The expression of PKR
showed that RV was able to replicate within the bronchial epithe- was significantly decreased in bronchial epithelium co-cultured
lium in both co-cultures during the first 24 h of infection. Interest- with asthmatic BSM cells and infected by RV after 24 h
ingly, the increase in the number of RV particles was significantly compared with that in bronchial epithelium co-cultured with
more pronounced in bronchial epithelium co-cultured with asth- non-asthmatic BSM cells at both the mRNA (Figure 2A) and pro-
matic BSM cells (Figures 1D and S1B). This result showed that tein (Figure 2B) levels. In another set of experiments, we also
asthmatic BSM cells were able to increase RV replication within observed a decreased PKR expression at 6 h, 24 h, and 48 h
the non-asthmatic bronchial epithelium. In another set of exper- post RV infection in bronchial epithelium co-cultured with asth-
iments, we obtained similar results using non-asthmatic or asth- matic BSM cells (Figure S5A). To validate these in vitro findings
matic bronchial epithelial cells co-cultured with non-asthmatic or ex vivo, we performed a proteomic comparison analysis of bron-
asthmatic BSM cells (Figure 1E). This result suggested that, in chial epithelial brushings obtained from either persons with
our co-culture model, the increase in RV replication was depen- asthma or non-asthmatic patients. We confirmed that the
dent on the asthmatic nature of the BSM cells but not that of the ex vivo protein expression of PKR was significantly decreased
bronchial epithelium. in asthmatic bronchial epithelium compared with that in non-
We also assessed interleukin (IL)-6 production by non-asth- asthmatic bronchial epithelium (Figure 2C).
matic bronchial epithelial cells co-cultured with asthmatic BSM
cells in response to RV infection using ELISA. Not surprisingly, Asthmatic BSM cells downregulated the PKR antiviral
RV infection induced an increase in IL-6 levels in bronchial epithe- pathway in bronchial epithelium
lial supernatant regardless of the co-culture conditions. However, Since double-stranded viral RNA, which is produced during RV
similar to what was observed for the number of RV particles, the replication, activates PKR by autophosphorylation at two major
level of IL-6 was further increased in the supernatant of RV-in- sites (Thr451 and Thr446) (Garcia et al., 2007), we thus assessed
fected non-asthmatic bronchial epithelium co-cultured with asth- the expression of phosphorylated PKR (P-PKR) in the bronchial
matic BSM cells compared with that in the supernatant of non- epithelium in vitro. We demonstrated that the levels of P-PKR
asthmatic bronchial epithelium co-cultured with non-asthmatic phosphorylated at Thr451 (Figure 3A) and Thr446 (Figure 3B)
BSM cells (Figure 1F). We then analyzed bronchial epithelial cell were both decreased in bronchial epithelium co-cultured with
apoptosis 24 h post RV infection by measuring the cleaved cas- asthmatic BSM cells and infected by RV for 24 h compared
pase 3 fluorescence. We did not find any difference in the level with those in bronchial epithelium co-cultured with non-asth-
of cell apoptosis in RV-infected bronchial epithelial cells co- matic BSM cells. In another set of experiments, the levels of
cultured with either non-asthmatic or asthmatic BSM cells (Fig- P-PKR phosphorylated at Thr451 were not altered at 6 h and
ure S3), providing that, in our experimental conditions (i.e., with 48 h post infection (Figure S5B) and those of P-PKR phosphory-
a MOI of 0.1 for 1 h as for all the other experiments), cell apoptosis lated at Thr446 were not detected at 6 h and 48 h post RV infec-
was not a possible mechanism of the increased RV replication. tion (Figure S5C). After its activation, PKR can phosphorylate the
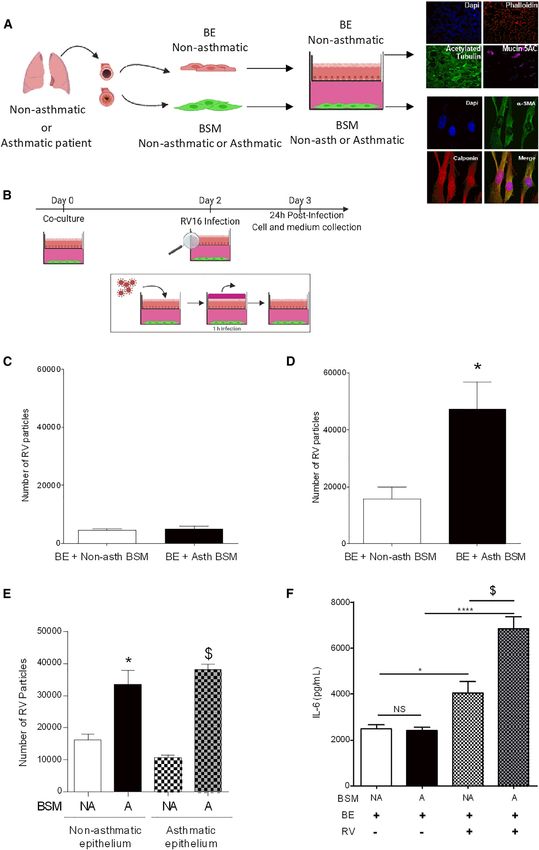
Figure 1. Asthmatic BSM increased RV replication in bronchial epithelium
(A) Schematic diagram of the co-culture model developed using differentiated air-liquid interface bronchial epithelium with ciliated and mucus cells on top of
bronchial smooth muscle (BSM) cells derived from either severe asthmatic or non-asthmatic persons. Confocal microscopy of reconstituted epithelium: nuclei
are stained with DAPI, actin with phalloidin, ciliated cells with acetylated tubulin, and mucus production with mucin 5AC. Confocal microscopy of BSM cells
stained with DAPI, a-SMA, and calponin.
(B) Schematic diagram of the co-culture model time lapse. Digital PCR of RV RNA particles.
(C and D) Non-asthmatic bronchial epithelial cells were co-cultured with either non-asthmatic (white bars, n = 9) or asthmatic (black bars, n = 7) BSM cells for
2 days before infection with RV (MOI 0.1). Digital PCR was performed after 6 h (C) or 24 h (D).
(E) Digital PCR of RV RNA particles. Non-asthmatic or asthmatic bronchial epithelium cells were co-cultured with either non-asthmatic (white bars, n = 4) or
asthmatic (black bars, n = 5) BSM cells for 2 days before infection for 24 h with RV (MOI: 0.1).
(F) Co-culture medium IL-6 concentration assessed by ELISA. Co-culture medium was collected from co-culture of bronchial epithelium with either non-asth-
matic (white bars, n = 4) or asthmatic BSM (black bars, n = 4), RV infected co-culture of bronchial epithelium with non-asthmatic BSM (squared white bars,
n = 4), asthmatic BSM (squared black bars, n = 4). Data are presented as mean ± SEM. *p < 0.05.
4 Cell Reports 38, 110571, March 29, 2022ll
Article OPEN ACCESS
Figure 2. Downregulation of PKR expression in asthmatic bronchial
epithelium
(A) Expression levels of PKR mRNA assessed by quantitative RT-PCR in
bronchial epithelial cells co-cultured with either non-asthmatic (white bars, n =
9) or asthmatic (black bars, n = 7) BSM cells for 2 days.
(B) Expression levels of PKR protein assessed by western blot in bronchial
epithelial cells co-cultured with either non-asthmatic (white bars, n = 5) or
asthmatic (black bars, n = 5) BSM cells for 2 days prior to 24 h of RV infection.
(C) Volcano plots of genes differentially expressed in ex vivo samples of
bronchial epithelium obtained from fibroscopic brushing of asthmatic (n = 3) or
non-asthmatic (n = 3) patients using mass spectrometry analysis. The y axis
line at y = 1.3 represents p = 0.05 of the fold change in asthmatic bronchial
epithelium. The x axis lines at x = 2.32 and x = 2.32 represent a fold change of
five times in asthmatic bronchial epithelium. Red dot represents PKR. Data are
presented as mean ± SEM. *p < 0.05.
initiation factor eIF2a at Ser51, which downregulates cellular
protein synthesis and consequently slows viral replication (Gar-
cia et al., 2007). Indeed, the level of P-eiF2a phosphorylated at
Ser51 assessed by western blotting (Figure 3C) or ELISA (Fig-
ure 3D) was also decreased in bronchial epithelium co-cultured
with asthmatic BSM cells compared with that in bronchial
epithelium co-cultured with non-asthmatic BSM cells. Moreover,
we also analyzed the global rate of protein synthesis using the
Click-iT HPG Alexa Fluor kit (Thermo Fisher Scientific) and
observed a slight but significantly decrease in protein translation
when bronchial epithelium was co-cultured with asthmatic BSM
cells (Figure 3E). Taken together, these results demonstrated
that asthmatic BSM cells increased both RV replication and
the IL-6 response in bronchial epithelium following RV infection
through impairment of the PKR antiviral pathway.
CCL20 expression was increased in asthmatic BSM
cells both in vitro and ex vivo
To explain the mechanism by which asthmatic BSM cells
decreased the activation of the PKR antiviral pathway and
increased RV replication within the bronchial epithelium, we
performed both unbiased transcriptomic and proteomic ana-
lyses at the BSM level (Tables S1 and S2). First, we compared
mRNA transcripts from asthmatic and non-asthmatic BSM cells
co-cultured with bronchial epithelium using NanoString technol-
ogy. Since BSM and bronchial epithelial cells were not in direct
contact in our model, we focused on secreted molecules over-
expressed by asthmatic BSM cells. CCL20 (i.e., macrophage
inflammatory protein-3alpha [MIP-3alpha]) mRNA appeared to
be highly overexpressed in asthmatic BSM cells compared
with non-asthmatic BSM cells (Figure 4A). Second, using prote-
omic analysis under the same experimental conditions, we
showed that the CCL20 protein was also overexpressed in
asthmatic BSM cells compared with non-asthmatic BSM cells
(Figure 4B). To confirm this finding, we analyzed the CCL20
level in the cell culture supernatant using ELISA. CCL20 was
nearly absent in supernatant from non-asthmatic or asthmatic
BSM cells cultured alone (Figure S6A), and it was expressed
at the basal level by bronchial epithelial cells cultured alone
(Figure 4C). However, CCL20 concentration within the superna-
tant of the bronchial epithelium co-cultured with both non-asth-
matic and asthmatic BSM cells was remarkably increased.
Moreover, CCL20 level was further increased in the supernatant
Cell Reports 38, 110571, March 29, 2022 5ll
OPEN ACCESS Article
Figure 3. Asthmatic BSM downregulated antiviral PKR and eIF2a activation in bronchial epithelium
(A–C) Expression levels of phospho-Thr 451 PKR (A), phospho-Thr 446 PKR (B), and phospho-Ser51 eIF2a (C) protein assessed by western blot in bronchial
epithelial cells co-cultured with either non-asthmatic (white bars, n = 5) or asthmatic (black bars, n = 5) BSM cells for 2 days prior to 24 h of RV infection.
(D) Quantification of phospho-Ser51 eIF2a by ELISA in bronchial epithelial cells co-cultured with either non-asthmatic (white bars, n = 7) or asthmatic (black bars,
n = 7) BSM cells for 2 days prior to 24 h of RV infection.
(E) Global rate of protein synthesis quantification assessed by fluorescence imaging in bronchial epithelial cells co-cultured with either non-asthmatic (white bars,
n = 7) or asthmatic (black bars, n = 5) BSM cells for 2 days prior to 24 h of RV infection. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01.
of bronchial epithelium co-cultured with asthmatic BSM cells co-cultured with bronchial epithelium. We observed an
compared with the supernatant of bronchial epithelium co- increased level of CCL20 expression in asthmatic BSM cells
cultured with non-asthmatic BSM cells (Figure 4C). We then compared with non-asthmatic cells (Figure 4D). Moreover, we
analyzed whether such an increase in CCL20 within the super- were also able to detect a significant increase in CCL20
natant was produced by BSM or bronchial epithelial cells. We mRNA in asthmatic BSM cells compared with non-asthmatic
observed the induction of CCL20 expression using ELISA on BSM cells after co-culture with bronchial epithelium (Figure 4E).
BSM lysates from both asthmatic and non-asthmatic persons In contrast, although bronchial epithelium was able to produce
6 Cell Reports 38, 110571, March 29, 2022ll
Article OPEN ACCESS
Figure 4. CCL20 expression was increased in
asthmatic BSM cells both in vitro and ex vivo
(A) Volcano plots of genes differentially expressed in
four asthmatic BSM cells compared with four non-
asthmatic BSM cells after 2 days of cell co-culture
with bronchial epithelial cells using NanoString
technology. The y axis line at y = 1.3 represents a
p = 0.05 of the fold change in asthmatic BSM cells.
The x axis lines at x = 2.32 and x = 2.32 represent a
fold change of five times in asthmatic BSM cells.
Red dot represents CCL20.
(B) Volcano plots of proteins differentially expressed
in three asthmatic BSM cells compared with three
non-asthmatic BSM cells after 2 days of cell co-cul-
ture with bronchial epithelial cells using mass spec-
trometry analysis. The y axis line at y = 1.3 represents
a p = 0.05 of the fold change in asthmatic BSM cells.
The x axis lines at x = 2.32 and x = 2.32 represent a
fold change of five times in asthmatic BSM cells. Red
dot represents CCL20. Proteomic data were clus-
tered into ‘‘respiratory virus’’ box pathway using in-
genuity pathway analysis (IPA).
(C) Co-culture medium CCL20 concentration as-
sessed by ELISA. Co-culture medium was collected
from bronchial epithelium only (gray bars, n = 4) and
co-culture of bronchial epithelium with either non-
asthmatic (white bars, n = 4) or asthmatic BSM
(black bars, n = 4).
(D) CCL20 concentration assessed by ELISA in
non-asthmatic (white bars, n = 5) or asthmatic
BSM (black bars, n = 5) cells co-cultured with bron-
chial epithelial cells.
(E) CCL20 mRNA expression assessed by RT-
qPCR assessed in non-asthmatic (white bars, n =
5) or asthmatic BSM (black bars, n = 5) cells co-
cultured with bronchial epithelial cells.
(F) Ex vivo CCL20 concentration assessed by
ELISA. Proteins were extracted from whole bi-
opsies of either non-asthmatic (white bars, n = 8)
or asthmatic (black bars, n = 6) patients. Results
were normalized by protein concentration of the
whole biopsies lysate. Data are presented as
mean ± SEM. *p < 0.05.
CCL20 inhibited the PKR antiviral
pathway and increased RV
replication in bronchial epithelium
To demonstrate the crucial role of CCL20 in
the bronchial epithelial PKR pathway, we
exposed bronchial epithelial cells to recom-
CCL20 at a basal level, we did not find any significant difference binant CCL20 in the absence of BSM cells for 48 h. We then
in the level of CCL20 produced by bronchial epithelium co- demonstrated that CCL20 significantly decreased both the PKR
cultured with either non-asthmatic or asthmatic BSM cells mRNA and protein levels in bronchial epithelial cells infected by
(Figure S6B). RV after 24 h (Figures 5A and 5B). In another set of experiments,
We also assessed the level of CCL20 ex vivo in patients’ bi- CCL20 did not alter the protein expression of PKR at 6 h and 48 h
opsies. Using whole bronchial biopsy lysates from non-asth- post RV infection (Figures S7A and S7B). Moreover, the activation
matic and asthmatic persons, we found a significant increase of the PKR antiviral pathway in bronchial epithelium was also
in CCL20 in the asthmatic biopsies (Figure 4F). Altogether, these decreased by CCL20 after RV infection. Indeed, CCL20 signifi-
results highlighted CCL20 as a potential candidate for specif- cantly inhibited the activation of PKR at both phosphorylation
ically decreasing the activation of the PKR antiviral pathway sites (i.e., Thr451 and Thr446) in bronchial epithelial cells infected
and increasing RV replication within the bronchial epithelium in by RV after 24 h (Figures 5C and 5D). The downstream PKR
the context of asthmatic BSM cell co-culture. signaling pathway has also been analyzed at different time points
Cell Reports 38, 110571, March 29, 2022 7ll
OPEN ACCESS Article
(legend on next page)
8 Cell Reports 38, 110571, March 29, 2022ll
Article OPEN ACCESS
post RV infection. We did not observe any activation of PKR at expression of Jnk in the bronchial epithelium (Figure 6D). We
both phosphorylation sites (i.e., Thr451 and Thr446) after 6 h or demonstrated that JNK silencing increased PKR expression in
48 h post RV infection (Figures S7A and S7B). The subsequent bronchial epithelial cells stimulated with CCL20 for 48 h (Figure 6E).
activation of eIF2a through Ser51 phosphorylation assessed by These results demonstrated that CCL20 inhibition of PKR expres-
both western blot (Figure 5E) and ELISA (Figure 5F) was sion is mediated by the activation of the Jnk pathway. Using the
decreased following CCL20 stimulation. Furthermore, the global same CRISPR-Cas9 technology, we successfully edited the
rate of protein synthesis was slightly but significantly decreased PKR gene (Figure 6F), and observed a significant decrease in
when bronchial epithelial cells were stimulated with CCL20 (Fig- PKR expression (Figure 6G) with a concomitant increase in RV par-
ure 5G). To explore the cellular pathway that caused CCL20 to ticle numbers (Figure 6H). These latter results confirmed the crucial
inhibit PKR, we first assessed the bronchial epithelial expression role of PKR below the CCL20/CCR6/Jnk activation pathway in
of its receptor, CCR6. We were able to detect CCR6 expression increasing RV replication within the bronchial epithelium.
on bronchial epithelium in vitro, whereas no differential expres-
sion was observed between bronchial epithelium co-cultured DISCUSSION
with non-asthmatic or asthmatic BSM cells (Figure S6C). Simi-
larly, no differential expression was observed between non-asth- Taken together, these results clearly demonstrated that BSM
matic and asthmatic bronchial epithelium ex vivo using western cells from persons with severe asthma increased RV replication
blot (data not shown). within the bronchial epithelium through an increase in CCL20
Finally, we analyzed RV replication in bronchial epithelium us- production by the BSM, which in turn activated the Jnk pathway
ing a CCL20 neutralizing antibody in the co-culture supernatant. and decreased the antiviral PKR/eIF2 pathway in the bronchial
Blocking CCL20 completely suppressed the increase in the RV epithelium.
particle number in bronchial epithelium co-cultured with asth- We have demonstrated a direct bottom-up effect of asthmatic
matic BSM cells, whereas the nonspecific antibody had no effect BSM on bronchial epithelium. Indeed, only the top-down effects
(Figure 5H). However, we were unable to demonstrate that block- of bronchial epithelium on BSM have previously been demon-
ing CCL20 rescued PKR activity (Figure S8). The role of CCL20 in strated. Indirectly, it has previously been shown that bronchial
RV infection of the bronchial epithelium was further confirmed, epithelium-derived factors, such as epithelial growth factor (Pan-
since CCL20 increased the number of RV RNA particles in the ettieri et al., 1996) or YKL-40 (Bara et al., 2012), induced both
bronchial epithelium (Figures 5I) 24 h post RV infection, without BSM cell proliferation and migration. Directly, Malavia et al.
modifications earlier at 6 h post RV infection (Figure S9). (2009) initially demonstrated that bronchial epithelium induced
To further explore the mechanisms of the effects of CCL20 on the proliferation of BSM cells from non-asthmatic subjects.
bronchial epithelium, we blocked the mitogen-activated protein ki- Furthermore, we previously showed that stimulation of the bron-
nase (MAPK), phosphoinositide 3-kinase (PI3K), or Janus kinase chial epithelium with house dust mite increased the proliferative
(Jnk) pathway using PD98059 (at 10 mM), wortmannin (at response of asthmatic BSM cells in a leukotriene-dependent
100 nM), or SP600125 (at 1 mM), respectively. These concentra- manner (Trian et al., 2015).
tions were selected since they correspond to the lower limit of their These results also suggested the cellular mechanisms by
range of activity used in the literature in bronchial epithelial cells to which asthmatic BSM played a central role in RV-induced
prevent side effects (Ip et al., 2007; Richard et al., 2018). Although asthma exacerbations. Analysis of both in vitro and ex vivo
both the MAPK and PI3K inhibitors had no effect (Figure S10), data clearly showed that asthmatic BSM cells decreased the
SP600125 significantly abolished the effect of CCL20 on PKR PKR response to RV infection in bronchial epithelium, which in
expression in bronchial epithelium (Figure 6A). We did confirm turn increased RV replication in bronchial epithelium. Despite
that SP600125 decreased the phosphorylation of Jnk (Figure 6B) the alteration of bronchial epithelium in asthma that was main-
in our experimental conditions (i.e., bronchial epithelial cells stim- tained in an air-liquid culture model (Gras et al., 2012), in our
ulated with CCL20 and infected by RV). In order to confirm the hands, co-culture with asthmatic bronchial epithelial cells did
implication of the Jnk pathway, we edited the JNK gene using not further improve RV replication induced by asthmatic BSM
CRISPR-Cas9 technology (Figure 6C), resulting in a decreased compared with that with non-asthmatic bronchial epithelium.
Figure 5. CCL20 downregulated antiviral PKR and eIF2a in bronchial epithelial cells resulting in increase in both RV replication and IL-6 pro-
duction
(A) PKR mRNA expression assessed by RT-qPCR. Bronchial epithelial cells were stimulated (+) or not () with 2 ng/mL of recombinant CCL20 (n = 4) for 2 days.
(B–E) Western blot full images and expression levels of PKR (B), phospho-Thr451 PKR (C), phospho-Thr446 PKR (D), and phospho-Ser51 eIF2a (E) proteins in
bronchial epithelial cells. Bronchial epithelial cells were stimulated (+) or not () with 2 ng/mL of recombinant CCL20 (n = 5) for 2 days prior to RV infection.
(F) Quantification of phospho-Ser51 eIF2a by ELISA in bronchial epithelial cells stimulated (+) or not () with 2 ng/mL of recombinant CCL20 (n = 6) for 2 days prior
to RV infection.
(G) Global rate of protein synthesis quantification assessed by florescence imaging in bronchial epithelial cells stimulated (+) or not () with 2 ng/mL of re-
combinant CCL20 (n = 6) for 2 days prior to RV infection.
(H) Digital PCR of RV RNA particles in bronchial epithelium after 24 h of RV infections. Bronchial epithelial cells were co-cultured with either non-asthmatic (white
bars, n = 5) or asthmatic (black bars, n = 5) BSM cells for 2 days before infection with RV (MOI 0.1). For CCL20 neutralization, the co-culture was either not treated
(open bars), or treated with anti-CCL20 blocking antibody (dotted bars) or nonspecific irrelevant antibody (IRR, hashed bars).
(I) Digital PCR of RV RNA particles in bronchial epithelial cells after 24 h of RV infections. Bronchial epithelial cells were stimulated (+) or not () with 2 ng/mL of
recombinant CCL20 (n = 5) for 2 days prior to RV infection. Data are mean ± SEM. *p < 0.05.
Cell Reports 38, 110571, March 29, 2022 9ll
OPEN ACCESS Article
Figure 6. CCL20 mediated Jnk pathway activation and downregulated antiviral PKR expression in bronchial epithelial cells, resulting in
increased RV particles number
(A) Expression levels of PKR protein were assessed by western blot in bronchial epithelial cells stimulated (+) (n = 8) or not () (n = 4), with 2 ng/mL of recombinant
CCL20, in the presence (+) or in the absence () of SP600125. Expression levels of Phospho-Jnk.
(legend continued on next page)
10 Cell Reports 38, 110571, March 29, 2022ll
Article OPEN ACCESS
These latter results may be considered to contradict previous been associated with reduced virus production, as demonstrated
studies, showing that asthmatic bronchial epithelium was char- in HeLa cells with an MOI of 20 (Foxman et al., 2016). Thus,
acterized by increased RV replication compared with controls apoptosis can be viewed as an alternative mechanism of antiviral
(Contoli et al., 2006; Wark et al., 2005). However, their experi- response. In the present study, we showed by contrast that RV-
mental conditions were different, with basal epithelial cells rather induced bronchial epithelial cell apoptosis was very low and
than fully differentiated epithelial cells in the present study. unchanged by asthmatic BSM cell co-culture. However, our
Indeed, recent studies performed with fully differentiated bron- experimental conditions used a very low MOI of 0.1. In addition,
chial epithelial cells did not identify any difference in RV replica- previous studies showed the crucial role of decreased interferon
tion between asthmatic and non-asthmatic cells (Jakiela et al., production by asthmatic bronchial epithelium in impairment of
2021; Veerati et al., 2020). Interestingly, the decreased expres- the RV response (Contoli et al., 2006; Zhu et al., 2019). However,
sion of PKR in the bronchial epithelium based on the ex vivo pro- PKR can also be activated by the interferon N-stimulated response
teomic data was not reproduced in air-liquid bronchial epithelial element (ISRE) present in the promoter region of its gene eIF2AK2
cells from asthmatic and non-asthmatic persons (data not (Kuhen and Samuel, 1997). Thus, the decreased PKR expression,
shown). This result suggested that the decrease in in vivo PKR found in the present study, could be an explanation in addition to
expression within bronchial epithelium is mediated by other interferon deficiency to explain the increased susceptibility to RV
cell types. The present in vitro results clearly demonstrated infection of persons with asthma. Although PKR has previously
that asthmatic BSM cells can play this role through increased been implicated in other diseases, such as Alzheimer disease
CCL20 production. However, various other cell types, such as and cancer (Garcia-Ortega et al., 2017), there is very little evidence
macrophages, mast cells, and dendritic cells, are known to pro- of the role of PKR in asthma. Only Loisel et al. (2016) identified a
duce high levels of CCL20 within the bronchial wall (Crane-God- single nuclear polymorphism in the eIF2AK2 gene, which encodes
reau et al., 2009; Homey et al., 2000; Marcet et al., 2007; Post PKR, that was significantly associated with asthma exacerbations
et al., 2013; Ramana et al., 2000). As a consequence, CCL20 or virus-induced wheezing phenotypes.
has previously been shown to be increased in asthmatic sputum To the best of our knowledge, the link between CCL20 and PKR
(Zijlstra et al., 2014) as well as in asthmatic BSM cells (Faiz et al., has never been demonstrated before. Here, we showed that PKR
2018), which was confirmed by the present study. Moreover, expression and function were downregulated by CCL20 in bron-
Faiz et al. (2018) demonstrated that CCL20 was able to increase chial epithelium. CCL20 specifically activated its unique receptor
bronchial epithelial mucus secretion. In addition, inhibition of CCR6 (Schutyser et al., 2003), but the mechanism leading to
CCL20 in a mouse model of asthma has been shown to enhance decreased PKR expression remains unknown. The CCR6 recep-
the antiviral response to RSV, through an attenuated recruitment tor belongs to the G protein-coupled receptor superfamily. The
of conventional dendritic cells (Kallal et al., 2010). Altogether, activation of the CCL20-CCR6 pathway has mostly been impli-
these previous findings clearly highlighted CCL20 as an impor- cated in cancer and inflammatory diseases. Indeed, CCL20 is
tant cytokine in asthma pathophysiology. In the present study, known to favor the chemotaxis of immune cells, leading to the
we provided evidence of the role of CCL20 in asthma exacerba- migration of Th17, regulatory T (Treg), B, and dendritic cells in can-
tions, since neutralizing CCL20 abrogated the increase in the RV cer, psoriasis, or rheumatoid arthritis (Brand et al., 2006; Lu et al.,
RNA particle number and the proinflammatory response in bron- 2018; Ranasinghe and Eri, 2018; Sullivan et al., 1999; Webb et al.,
chial epithelium co-cultured with asthmatic BSM cells. Thus, we 2008; Weckmann et al., 2007; Zhang et al., 2017). In previous pub-
can speculate that CCL20 blockade may be used in asthma ther- lications, the authors demonstrated that the downstream
apy by altering the balance of innate immune cells (Kallal et al., signaling pathways of CCR6 activation could be associated with
2010), and mediating an efficient antiviral response to viruses, the PI3-AKT, MAPK-ERK, or even MAPK-Jnk pathways. In the
including RV, at the bronchial epithelium level. To date, there present study, using a specific MAPK-Jnk pathway inhibitor
are several CCL20 or CCR6 inhibitors that are in early phases (SP600125) and dedicated CRISPR-Cas9 gene editing, we
of development in cancers and other inflammatory diseases showed that CCL20-CCR6 led to activation of the MAPK-Jnk
(Brand et al., 2006; Lu et al., 2018; Ranasinghe and Eri, 2018; pathway within bronchial epithelial cells. The MAPK-Jnk pathway
Sullivan et al., 1999; Webb et al., 2008; Weckmann et al., 2007; is commonly associated with the activation of transcription factors
Zhang et al., 2017). However, it is difficult to predict their effi- such as STATs, which have been described as activators or re-
cacy/tolerance ratio in asthma patients. pressors of gene expression (Ramana et al., 2000). When we
The present study may appear controversial compared with analyzed the sequence of the eIF2AK2 gene using a human
previous studies. Indeed, cell apoptosis in RV-infected cells has gene database (GeneCards), we found potential transcription
(B) Proteins were assessed by western blot in bronchial epithelial cells stimulated with 2 ng/mL of recombinant CCL20, in the presence (+) or in the absence () of
SP600125 (n = 7) for 2 days prior to RV infection.
(C) The presence of JNK was assessed by PCR in bronchial epithelial cells edited for JNK gene.
(D and E) Expression levels of Jnk (D) and PKR (E) proteins were assessed by western blot in bronchial epithelial cells either non-edited (white squares, n = 6) or
edited for JNK gene (black circles, n = 6).
(F) The presence of PKR was assessed by PCR in bronchial epithelial cells edited for PKR gene.
(G) Expression levels of PKR proteins were assessed by western blot in bronchial epithelial cells either non-edited (white squares, n = 6) or edited for PKR gene
(black circles, n = 6).
(H) Digital PCR of RV RNA particles in bronchial epithelial cells either non-edited (white squares, n = 6) or edited for PKR gene (black circles, n = 6) after 24 h of RV
infections. Data are mean ± SEM. *p < 0.05.
Cell Reports 38, 110571, March 29, 2022 11ll
OPEN ACCESS Article
factors, such as SP proteins, which were already shown in the B Data and code availability
literature to regulate PKR expression (Das et al., 2006), and d EXPERIMENTAL MODEL AND SUBJECT DETAILS
GATA, FOXO, SOX, and STAT proteins, which are capable of as- d METHOD DETAILS
sociation with the regulatory region of the promoter. These obser- B Cell culture
vations, which are associated with the effects of CCL20 on B Co-culture model
decreasing PKR mRNA in bronchial epithelial cells, led us to hy- B Immunocytochemistry of differentiated bronchial
pothesize that CCL20 activates or represses transcription factors, epithelial and BSM cells
leading to the inhibition of PKR gene expression. B Rhinovirus production and infection
B Cell apoptosis
Limitations of the study B ELISA
Several limitations can be discussed in relation to this study. B Transcriptomic analysis (NanoString)
First, as mentioned above, BSM was not the only CCL20 pro- B Western blotting
ducer. However, CCL20 production by other cell types was B RNA extraction and real-time quantitative PCR (qPCR)
induced after epithelial stimulation. In our model, the epithelial B Digital PCR
production of CCL20 was much lower than that observed in B Measurement of the global rate of protein synthesis
the co-culture supernatant and was not induced by BSM co-cul- B CRISPR-based gene targeting
ture. In this study, we clearly highlighted the role of CCL20 in the B Proteomic analysis
PKR antiviral pathway in bronchial epithelium and the ability of d QUANTIFICATION AND STATISTICAL ANALYSIS
asthmatic BSM cells to substantially produce this cytokine
when co-cultured with bronchial epithelium. Moreover, the role SUPPLEMENTAL INFORMATION
of BSM cells in CCL20 production within the bronchus could
Supplemental information can be found online at https://doi.org/10.1016/j.
be greater in persons with asthma, since the increase in BSM celrep.2022.110571.
mass in those patients is significant (Trian et al., 2007) and the
distance between the BSM and bronchial epithelium is ACKNOWLEDGMENTS
decreased (Pepe et al., 2005). Second, we only used human
samples both ex vivo and in vitro. Thus, we did not confirm our We thank the staffs of both the pathology and surgery departments (both from
the University Hospital of Bordeaux), Isabelle Goasdoue, Virginie Niel, and Ma-
findings in vivo using an animal model of asthma. However,
rine Servat from the clinical investigation center, for technical assistance.
although animal models have been widely used in the literature, Regarding project funding, we strongly acknowledge the Fondation Thierry
their results cannot be extrapolated systematically to human dis- et Annick Desmarest, du Fonds de Dotation Recherche en Santé Respiratoire
eases. In particular, the use of RV16 is not possible in mice since et de la Fondation du Souffle, the Fondation de l’Université de Bordeaux for the
the ICAM-1 receptor is not expressed by murine bronchial FGLMR/AVAD funding, the Fondation pour la Recherche Médicale, the
epithelial cells (Bartlett et al., 2008). Moreover, to the best of Agence Nationale de la Recherche, and an unrestricted grant from
our knowledge, there is no real animal model of severe asthma, AstraZeneca.
Sources of support: the project was funded by the Fondation Thierry et An-
particularly non-allergic severe asthma. Third, we were unable to
nick Desmarest, du Fonds de Dotation Recherche en Santé Respiratoire et de
demonstrate that blocking CCL20 was able to rescue PKR pro- la Fondation du Souffle (lauréat de l’appel d’offres commun 2015 FD2015),
tein expression, whereas anti-CCL20 decreased RV particles Fondation de l’Université de Bordeaux (Fonds pour les maladies chroniques
numbers on the one hand, and, on the other hand, recombinant nécessitant une assistance médico-technique FGLMR/AVAD), Fondation
CCL20 decreased PKR protein expression and phosphorylation pour la Recherche Médicale (DEQ20170336706), Agence Nationale de la Re-
and increased RV particle numbers. cherche (ANR, ROSAE project CE14-0015-01), and an unrestricted grant from
AstraZeneca.
In conclusion, this study demonstrated the direct bottom-up
effect of BSM from persons with severe asthma on the suscep- AUTHOR CONTRIBUTIONS
tibility of bronchial epithelium to RV infection. Indeed, severe
asthmatic BSM cells decreased the efficiency of the PKR P.E. performed the vast majority of the in vitro and ex vivo experiments and
pathway in bronchial epithelium after RV infection, which was wrote the first draft of the manuscript. B.A. and A.C. helped to perform both
associated with an increased secretion of CCL20. The identified the in vitro and ex vivo experiments. E.M. and O.O. technically assisted with
BSM-to-bronchial epithelium bottom-up axis is of high interest in the in vitro experiments. M.T., P.O.G., and P.B. conducted patient recruitment.
T.L.-L. performed the digital PCR experiments. J.-W.D. performed the prote-
understanding and preventing RV-induced asthma exacerba-
omics experiments. I.D., B.A., and R.M. helped to design the study and edited
tion, which remains a major unmet need for severe asthma. the manuscript. P.E., T.T., and P.B. conceived the project, designed and su-
pervised the study, analyzed the data, and edited the manuscript.
STAR+METHODS
DECLARATION OF INTERESTS
Detailed methods are provided in the online version of this paper
- P.B. reports grants and personal fees from Novartis; grants, personal fees,
and include the following: and non-financial support from Boehringer Ingelheim; personal fees and
non-financial support from Chiesi, AstraZeneca, and Sanofi; personal
d KEY RESOURCES TABLE
fees from Menarinni, and TEVA, outside the submitted work. In addition,
d RESOURCE AVAILABILITY he has a delivered patent (EP N 15152886.6; i.e., New Compositions
B Lead contact and Methods of Treating and/or Preventing Chronic Obstructive Pulmo-
B Materials availability nary Disease), a submitted patent (22605-FR; i.e., Geometric
12 Cell Reports 38, 110571, March 29, 2022ll
Article OPEN ACCESS
Characterization of Airways using MRI), and a submitted patent (EP of deficient type III interferon-lambda production in asthma exacerbations.
N 20173595.8; i.e., New Compositions and Methods of Treating COVID- Nat. Med. 12, 1023–1026.
19 Disease), all outside the submitted work. Cox, G., Thomson, N.C., Rubin, A.S., Niven, R.M., Corris, P.A., Siersted, H.C.,
- P.-O.G. reports research support from AstraZeneca outside the submit- Olivenstein, R., Pavord, I.D., McCormack, D., Chaudhuri, R., et al. (2007).
ted work. In addition, he has a delivered patent (EP 15152886.6; i.e., New Asthma control during the year after bronchial thermoplasty. N. Engl. J.
Compositions and Methods of Treating and/or Preventing Chronic Med. 356, 1327–1337.
Obstructive Pulmonary Disease) and a submitted patent (EP
Crane-Godreau, M.A., Maccani, M.A., Eszterhas, S.K., Warner, S.L., Jukosky,
N 20173595.8; i.e., New Compositions and Methods of Treating
J.A., and Fiering, S. (2009). Exposure to cigarette smoke disrupts CCL20-
COVID-19 Disease), all outside the submitted work.
mediated antimicrobial activity in respiratory epithelial cells. Open Immunol.
- I..D. has a delivered patent (EP 15152886; i.e., New Compositions and
J. 2, 86–93.
Methods of Treating and/or preventing Chronic Obstructive Pulmonary
Disease), and a submitted patent (EP N 20173595.8; i.e., New Compo- Das, S., Ward, S.V., Tacke, R.S., Suske, G., and Samuel, C.E. (2006). Activa-
sitions and Methods of Treating COVID-19 Disease), all outside the sub- tion of the RNA-dependent protein kinase PKR promoter in the absence of
mitted work. interferon is dependent upon Sp proteins. J. Biol. Chem. 281, 3244–3253.
- All other authors declare no competing interests. Faiz, A., Weckmann, M., Tasena, H., Vermeulen, C.J., Van den Berge, M., Ten
Hacken, N.H.T., Halayko, A.J., Ward, J.P.T., Lee, T.H., Tjin, G., et al. (2018).
Received: January 18, 2021 Profiling of healthy and asthmatic airway smooth muscle cells following inter-
Revised: December 13, 2021 leukin-1beta treatment: a novel role for CCL20 in chronic mucus hypersecre-
Accepted: March 4, 2022 tion. Eur. Respir. J. 52, 1800310.
Published: March 29, 2022 Foxman, E.F., Storer, J.A., Vanaja, K., Levchenko, A., and Iwasaki, A. (2016).
Two interferon-independent double-stranded RNA-induced host defense
REFERENCES strategies suppress the common cold virus at warm temperature. Proc. Natl.
Acad. Sci. U S A. 113, 8496–8501.
Bara, I., Ozier, A., Girodet, P.O., Carvalho, G., Cattiaux, J., Begueret, H., Thu-
Garcia-Ortega, M.B., Lopez, G.J., Jimenez, G., Garcia-Garcia, J.A., Conde, V.,
merel, M., Ousova, O., Kolbeck, R., Coyle, A.J., et al. (2012). Role of YKL-40 in
Boulaiz, H., Carrillo, E., Peran, M., Marchal, J.A., and Garcia, M.A. (2017). Clin-
bronchial smooth muscle remodeling in asthma. Am. J. Respir. Crit. Care Med.
ical and therapeutic potential of protein kinase PKR in cancer and metabolism.
185, 715–722.
Expert Rev. Mol. Med. 19, e9.
Bartlett, N.W., Walton, R.P., Edwards, M.R., Aniscenko, J., Caramori, G., Zhu,
Garcia, M.A., Meurs, E.F., and Esteban, M. (2007). The dsRNA protein kinase
J., Glanville, N., Choy, K.J., Jourdan, P., Burnet, J., et al. (2008). Mouse models
PKR: virus and cell control. Biochimie 89, 799–811.
of rhinovirus-induced disease and exacerbation of allergic airway inflamma-
tion. Nat. Med. 14, 199–204. Gielen, V., Sykes, A., Zhu, J., Chan, B., Macintyre, J., Regamey, N., Kieninger,
Bateman, E.D., Hurd, S.S., Barnes, P.J., Bousquet, J., Drazen, J.M., FitzGer- E., Gupta, A., Shoemark, A., Bossley, C., et al. (2015). Increased nuclear sup-
ald, J.M., Gibson, P., Ohta, K., O’Byrne, P., Pedersen, S.E., Pizzichini, E., et al. pressor of cytokine signaling 1 in asthmatic bronchial epithelium suppresses
(2018). Global strategy for asthma management and prevention: GINA execu- rhinovirus induction of innate interferons. J. Allergy Clin. Immunol. 136, 177–
tive summary. Eur. Respir. J. 31, 143–178. 188.e11.
Bel, E.H., Wenzel, S.E., Thompson, P.J., Prazma, C.M., Keene, O.N., Yancey, Girkin, J., Hatchwell, L., Foster, P., Johnston, S.L., Bartlett, N., Collison, A.,
S.W., Ortega, H.G., Pavord, I.D., and Investigators, S. (2014). Oral glucocorti- and Mattes, J. (2015). CCL7 and IRF-7 mediate hallmark inflammatory and
coid-sparing effect of mepolizumab in eosinophilic asthma. N. Engl. J. Med. IFN responses following rhinovirus 1B infection. J. Immunol. 194, 4924–4930.
371, 1189–1197. Girodet, P.O., Allard, B., Thumerel, M., Begueret, H., Dupin, I., Ousova, O.,
Bleecker, E.R., FitzGerald, J.M., Chanez, P., Papi, A., Weinstein, S.F., Barker, Lassalle, R., Maurat, E., Ozier, A., Trian, T., et al. (2016). Bronchial smooth
P., Sproule, S., Gilmartin, G., Aurivillius, M., Werkstrom, V., et al. (2016). Effi- muscle remodeling in nonsevere asthma. Am. J. Respir. Crit. Care Med.
cacy and safety of benralizumab for patients with severe asthma uncontrolled 193, 627–633.
with high-dosage inhaled corticosteroids and long-acting beta2-agonists Girodet, P.O., Dournes, G., Thumerel, M., Begueret, H., Dos Santos, P., Ozier,
(SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lan- A., Dupin, I., Trian, T., Montaudon, M., Laurent, F., et al. (2015). Calcium chan-
cet 388, 2115–2127. nel blocker reduces airway remodeling in severe asthma: a proof-of-concept
Brand, S., Olszak, T., Beigel, F., Diebold, J., Otte, J.M., Eichhorst, S.T., Goke, study. Am. J. Respir. Crit. Care Med. 191, 876–883.
B., and Dambacher, J. (2006). Cell differentiation dependent expressed CCR6 Gras, D., Bourdin, A., Vachier, I., de Senneville, L., Bonnans, C., and Chanez,
mediates ERK-1/2, SAPK/JNK, and Akt signaling resulting in proliferation and P. (2012). An ex vivo model of severe asthma using reconstituted human bron-
migration of colorectal cancer cells. J. Cell Biochem. 97, 709–723. chial epithelium. J. Allergy Clin. Immunol. 129, 1259–1266.e51.
Busse, W.W., Lemanske, R.F., Jr., and Gern, J.E. (2010). Role of viral respira- Henriet, E., Abou Hammoud, A., Dupuy, J.W., Dartigues, B., Ezzoukry, Z., Du-
tory infections in asthma and asthma exacerbations. Lancet 376, 826–834. got-Senant, N., Leste-Lasserre, T., Pallares-Lupon, N., Nikolski, M., Le Bail,
Cakebread, J.A., Xu, Y., Grainge, C., Kehagia, V., Howarth, P.H., Holgate, S.T., B., et al. (2017). Argininosuccinate synthase 1 (ASS1): a marker of unclassified
and Davies, D.E. (2011). Exogenous IFN-beta has antiviral and anti-inflamma- hepatocellular adenoma and high bleeding risk. Hepatology 66, 2016–2028.
tory properties in primary bronchial epithelial cells from asthmatic subjects Homey, B., Dieu-Nosjean, M.C., Wiesenborn, A., Massacrier, C., Pin, J.J., Old-
exposed to rhinovirus. J. Allergy Clin. Immunol. 127, 1148–1154.e49. ham, E., Catron, D., Buchanan, M.E., Muller, A., deWaal Malefyt, R., et al.
Castro, M., Corren, J., Pavord, I.D., Maspero, J., Wenzel, S., Rabe, K.F., (2000). Up-regulation of macrophage inflammatory protein-3 alpha/CCL20
Busse, W.W., Ford, L., Sher, L., FitzGerald, J.M., et al. (2018). Dupilumab effi- and CC chemokine receptor 6 in psoriasis. J. Immunol. 164, 6621–6632.
cacy and safety in moderate-to-severe uncontrolled asthma. N. Engl. J. Med. Humbert, M., Beasley, R., Ayres, J., Slavin, R., Hebert, J., Bousquet, J., Beeh,
378, 2486–2496. K.M., Ramos, S., Canonica, G.W., Hedgecock, S., et al. (2005). Benefits of
Chung, K.F., Wenzel, S.E., Brozek, J.L., Bush, A., Castro, M., Sterk, P.J., Ad- omalizumab as add-on therapy in patients with severe persistent asthma
cock, I.M., Bateman, E.D., Bel, E.H., Bleecker, E.R., et al. (2014). International who are inadequately controlled despite best available therapy (GINA 2002
ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. step 4 treatment): INNOVATE. Allergy 60, 309–316.
Eur. Respir. J. 43, 343–373. Ip, W.K., Wong, C.K., Li, M.L., Li, P.W., Cheung, P.F., and Lam, C.W. (2007).
Contoli, M., Message, S.D., Laza-Stanca, V., Edwards, M.R., Wark, P.A., Bar- Interleukin-31 induces cytokine and chemokine production from human bron-
tlett, N.W., Kebadze, T., Mallia, P., Stanciu, L.A., Parker, H.L., et al. (2006). Role chial epithelial cells through activation of mitogen-activated protein kinase
Cell Reports 38, 110571, March 29, 2022 13ll
OPEN ACCESS Article
signalling pathways: implications for the allergic response. Immunology 122, Ramana, C.V., Chatterjee-Kishore, M., Nguyen, H., and Stark, G.R. (2000).
532–541. Complex roles of Stat1 in regulating gene expression. Oncogene 19, 2619–
Jakiela, B., Rebane, A., Soja, J., Bazan-Socha, S., Laanesoo, A., Plutecka, H., 2627.
Surmiak, M., Sanak, M., Sladek, K., and Bochenek, G. (2021). Remodeling of Ranasinghe, R., and Eri, R. (2018). Modulation of the CCR6-CCL20 Axis: a po-
bronchial epithelium caused by asthmatic inflammation affects its response to tential therapeutic target in inflammation and cancer. Medicina (Kaunas) 54,
rhinovirus infection. Sci. Rep. 11, 12821. 88.
Kallal, L.E., Schaller, M.A., Lindell, D.M., Lira, S.A., and Lukacs, N.W. (2010). Richard, N., Marti, L., Varrot, A., Guillot, L., Guitard, J., Hennequin, C., Imberty,
CCL20/CCR6 blockade enhances immunity to RSV by impairing recruitment A., Corvol, H., Chignard, M., and Balloy, V. (2018). Human bronchial epithelial
of DC. Eur. J. Immunol. 40, 1042–1052. cells inhibit Aspergillus fumigatus germination of extracellular conidia via FleA
Kaminska, M., Foley, S., Maghni, K., Storness-Bliss, C., Coxson, H., Ghezzo, recognition. Sci. Rep. 8, 15699.
H., Lemiere, C., Olivenstein, R., Ernst, P., Hamid, Q., et al. (2009). Airway re- Schutyser, E., Struyf, S., and Van Damme, J. (2003). The CC chemokine
modeling in subjects with severe asthma with or without chronic persistent CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 14, 409–426.
airflow obstruction. J. Allergy Clin. Immunol. 124, 45–51. Sullivan, S.K., McGrath, D.A., Liao, F., Boehme, S.A., Farber, J.M., and Bacon,
Kim, T.K., Bheda-Malge, A., Lin, Y., Sreekrishna, K., Adams, R., Robinson, K.B. (1999). MIP-3alpha induces human eosinophil migration and activation of
M.K., Bascom, C.C., Tiesman, J.P., Isfort, R.J., and Gelinas, R. (2015). A sys- the mitogen-activated protein kinases (p42/p44 MAPK). J. Leukoc. Biol. 66,
tems approach to understanding human rhinovirus and influenza virus infec- 674–682.
tion. Virology 486, 146–157. To, T., Stanojevic, S., Moores, G., Gershon, A.S., Bateman, E.D., Cruz, A.A.,
Koh, K.D., Siddiqui, S., Cheng, D., Bonser, L.R., Sun, D.I., Zlock, L.T., Fink- and Boulet, L.P. (2012). Global asthma prevalence in adults: findings from
beiner, W.E., Woodruff, P.G., and Erle, D.J. (2020). Efficient RNP-directed hu- the cross-sectional world health survey. BMC Public Health 12, 204.
man gene targeting reveals SPDEF is required for IL-13-induced mucostasis. Trian, T., Allard, B., Dupin, I., Carvalho, G., Ousova, O., Maurat, E., Bataille, J.,
Am. J. Respir. Cell Mol. Biol. 62, 373–381. Thumerel, M., Begueret, H., Girodet, P.O., et al. (2015). House dust mites
Kuhen, K.L., and Samuel, C.E. (1997). Isolation of the interferon-inducible induce proliferation of severe asthmatic smooth muscle cells via an epithe-
RNA-dependent protein kinase Pkr promoter and identification of a novel lium-dependent pathway. Am. J. Respir. Crit. Care Med. 191, 538–546.
DNA element within the 5’-flanking region of human and mouse Pkr genes. Trian, T., Benard, G., Begueret, H., Rossignol, R., Girodet, P.O., Ghosh, D.,
Virology 227, 119–130. Ousova, O., Vernejoux, J.M., Marthan, R., Tunon-de-Lara, J.M., et al. (2007).
Kuo, C., Lim, S., King, N.J.C., Johnston, S.L., Burgess, J.K., Black, J.L., and Bronchial smooth muscle remodeling involves calcium-dependent enhanced
Oliver, B.G. (2011). Rhinovirus infection induces extracellular matrix protein mitochondrial biogenesis in asthma. J. Exp. Med. 204, 3173–3181.
deposition in asthmatic and nonasthmatic airway smooth muscle cells. Am. Trian, T., Moir, L.M., Ge, Q., Burgess, J.K., Kuo, C., King, N.J., Reddel, H.K.,
J. Physiol. Lung Cell Mol. Physiol. 300, L951–L957. Black, J.L., Oliver, B.G., and McParland, B.E. (2010). Rhinovirus-induced ex-
Loisel, D.A., Du, G., Ahluwalia, T.S., Tisler, C.J., Evans, M.D., Myers, R.A., acerbations of asthma: how is the {beta}2-adrenoceptor implicated? Am. J.
Gangnon, R.E., Kreiner-Moller, E., Bonnelykke, K., Bisgaard, H., et al. Respir. Cell Mol. Biol. 43, 227–233.
(2016). Genetic associations with viral respiratory illnesses and asthma control Veerati, P.C., Troy, N.M., Reid, A.T., Li, N.F., Nichol, K.S., Kaur, P., Maltby, S.,
in children. Clin. Exp. Allergy 46, 112–124. Wark, P.A.B., Knight, D.A., Bosco, A., et al. (2020). Airway epithelial cell immu-
Lu, M.Y., Lu, S.S., Chang, S.L., and Liao, F. (2018). The phosphorylation of nity is delayed during rhinovirus infection in asthma and COPD. Front. Immu-
CCR6 on distinct ser/Thr residues in the carboxyl Terminus differentially regu- nol. 11, 974.
lates biological function. Front. Immunol. 9, 415. Wark, P.A., Johnston, S.L., Bucchieri, F., Powell, R., Puddicombe, S., Laza-
Malavia, N.K., Raub, C.B., Mahon, S.B., Brenner, M., Panettieri, R.A., Jr., and Stanca, V., Holgate, S.T., and Davies, D.E. (2005). Asthmatic bronchial epithe-
George, S.C. (2009). Airway epithelium stimulates smooth muscle prolifera- lial cells have a deficient innate immune response to infection with rhinovirus.
tion. Am. J. Respir. Cell Mol. Biol. 41, 297–304. J. Exp. Med. 201, 937–947.
Marcet, B., Horckmans, M., Libert, F., Hassid, S., Boeynaems, J.M., and Com- Webb, A., Johnson, A., Fortunato, M., Platt, A., Crabbe, T., Christie, M.I., Watt,
muni, D. (2007). Extracellular nucleotides regulate CCL20 release from human G.F., Ward, S.G., and Jopling, L.A. (2008). Evidence for PI-3K-dependent
primary airway epithelial cells, monocytes and monocyte-derived dendritic migration of Th17-polarized cells in response to CCR2 and CCR6 agonists.
cells. J. Cell Physiol. 211, 716–727. J. Leukoc. Biol. 84, 1202–1212.
Ortega, H.G., Liu, M.C., Pavord, I.D., Brusselle, G.G., FitzGerald, J.M., Chetta, Wechsler, M.E., Laviolette, M., Rubin, A.S., Fiterman, J., Lapa e Silva, J.R.,
A., Humbert, M., Katz, L.E., Keene, O.N., Yancey, S.W., et al. (2014). Mepoli- Shah, P.L., Fiss, E., Olivenstein, R., Thomson, N.C., Niven, R.M., et al.
zumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. (2013). Bronchial thermoplasty: long-term safety and effectiveness in patients
371, 1198–1207. with severe persistent asthma. J. Allergy Clin. Immunol. 132, 1295–1302.
Panettieri, R.A., Jr., Goldie, R.G., Rigby, P.J., Eszterhas, A.J., and Hay, D.W. Weckmann, M., Collison, A., Simpson, J.L., Kopp, M.V., Wark, P.A., Smyth,
(1996). Endothelin-1-induced potentiation of human airway smooth muscle M.J., Yagita, H., Matthaei, K.I., Hansbro, N., Whitehead, B., et al. (2007). Crit-
proliferation: an ETA receptor-mediated phenomenon. Br. J. Pharmacol. ical link between TRAIL and CCL20 for the activation of TH2 cells and the
118, 191–197. expression of allergic airway disease. Nat. Med. 13, 1308–1315.
Pepe, C., Foley, S., Shannon, J., Lemiere, C., Olivenstein, R., Ernst, P., Ludwig, Zhang, X.P., Hu, Z.J., Meng, A.H., Duan, G.C., Zhao, Q.T., and Yang, J. (2017).
M.S., Martin, J.G., and Hamid, Q. (2005). Differences in airway remodeling be- Role of CCL20/CCR6 and the ERK signaling pathway in lung adenocarcinoma.
tween subjects with severe and moderate asthma. J. Allergy Clin. Immunol. Oncol. Lett. 14, 8183–8189.
116, 544–549. Zhu, J., Message, S.D., Mallia, P., Kebadze, T., Contoli, M., Ward, C.K., Bar-
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., nathan, E.S., Mascelli, M.A., Kon, O.M., Papi, A., et al. (2019). Bronchial
Kundu, D.J., Inuganti, A., Griss, J., Mayer, G., Eisenacher, M., et al. (2019). The mucosal IFN-alpha/beta and pattern recognition receptor expression in pa-
PRIDE database and related tools and resources in 2019: improving support tients with experimental rhinovirus-induced asthma exacerbations. J. Allergy
for quantification data. Nucleic Acids Res. 47, D442–D450. Clin. Immunol. 143, 114–125.e14.
Post, S., Nawijn, M.C., Jonker, M.R., Kliphuis, N., van den Berge, M., van Oos- Zijlstra, G.J., Fattahi, F., Rozeveld, D., Jonker, M.R., Kliphuis, N.M., van den
terhout, A.J., and Heijink, I.H. (2013). House dust mite-induced calcium Berge, M., Hylkema, M.N., ten Hacken, N.H., van Oosterhout, A.J., and Heijink,
signaling instigates epithelial barrier dysfunction and CCL20 production. Al- I.H. (2014). Glucocorticoids induce the production of the chemoattractant
lergy 68, 1117–1125. CCL20 in airway epithelium. Eur. Respir. J. 44, 361–370.
14 Cell Reports 38, 110571, March 29, 2022You can also read

















































