AQUATIC MICROBIAL AND MOLECULAR ECOLGY - 2021 LABORATORY MANUAL Department of Biology University of Southern Denmark
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
AQUATIC MICROBIAL AND
MOLECULAR ECOLGY
2021
LABORATORY MANUAL
Department of Biology
University of Southern DenmarkWorkplan for AMME 2021
(AT-Alexander Treusch, BT- Bo Thamdrup, DEC - Don E Canfield, RNG – Ronnie N Glud)
Sun 15.08: 14:00 – 18:00 Excursion to field sites and collection of sediment & water
RNG+AT
18:00 -- Icebreaker barbecue (ALL?)
Mon 16.08 8.30 – 9:00 Introduction – RNG
9:15 – 10:00 Lecture; The role of prokaryote diversity - AT
10:30 – 12:00 Laboratory work
13:00 – 17:00 Laboratory work
Tues 17.08 8:30 – 10:00 Lecture; Structure & growth of microbial populations - AT
10:30 – 12:00 Laboratory work
13:00 – 17:00 Laboratory work
Wed 18.08 8:30 – 10:00 Lecture; Microbial energetics - RNG
10:30 – 12:00 Laboratory work
13:00 – 17:00 Laboratory work
Thur 19.08 8:30 – 10:00 Lecture; Oxygen cycling & dynamics – RNG
10:30 – 12:00 Oral student presentations
13:00 – 17:00 Laboratory work
Fri 20.08 8:30 – 10:00 Lecture; The biogeochemistry of nitrogen - BT
10:30 – 12:00 Laboratory work
13:00 – 17:00 Laboratory work
Sat 21.08 8:30 – 10:00 Lecture; The biogeochemistry of methane - BT
10:30 – 12:00 Oral student presentations
13:00 – 17:00 Laboratory work
Sun 22.08 Off or/and additional sediment sampling
Mon 23.08 8:30 – 10:00 Lecture; Carbon cycling - RNG
10:30 – 12:00 Laboratory work
13:00 – 17:00 Laboratory work
2Tues 24.08 8:30 – 10:00 Lecture; The biogeochemistry of Sulfur - DEC
10:30 – 12:00 Laboratory work
13:00 – 17:00 Laboratory work
Wed 25.08 8:30 – 10:00 Lecture; The biogeochemistry of Iron and manganese - DEC
10:30 – 12:00 Laboratory work
13:00 – 17:00 Laboratory work
Thur 26.08 8:30 – 10:00 Lecture; Molecular microbial ecology - AT
10:30 – 12:00 Laboratory work
13:00 – 17:00 Laboratory work
Fri 27.08 8:30 – 12:00 Student Data presentation
13:00 – 16:00 Report writing & discussions
Sat 28.08 8:30 – 12:00 Report writing & discussions
13:00 – 16:00 Report writing & discussions
18.00 -- Joint dinner
Sun 29.08 8:30 – 12:00 Report writing & discussions
13:00 – 13:15 Group photo
13:15 – 16:00 Report writing & discussions
3EACH TEAM’S TIME SCHEDULE FOR THE VARIOUS EXERCISES
The codes refer to chapters in the ”Procedures” section of each exercise.
KF indicates Kærby Fed, FS indicates Fællesstrand,
Date Team 1 Team 2 Team 3 Team 4 Team 5
KF KF KF FS FS
Mon 16.08 1A 1A 1A 1A 1A
10:30-17 7 7 7 7 7
8ABC 8ABC 8ABC 8ABC 8ABC
Tue 17.08 1B 1B 1B 1B 1B
10:30-17 2ABCD 4AB 6AB 4AB 5.1 & 5.2
Wed 18.08 1C 1C 1CDI 1C 1C
10:30-17 4AB 2ABCD 4CDE 6AB
Thu 19.08 6AB 2E 1DII 5.1 & 5.2 1DI
13-17 2ABCD
Fri 20.08 4CDE 1DI 2E 2ABCD 1DII
10:30-17 4AB 3AB
Sat 21.08 1DI 1DII 3AB 2E 4AB
13-17 5.1A & 5.2 6AB
Mon 23.08 1DII 4CDE 5.1 & 5.2 3AB 2ABCD
10:30-17
Tue 24.08 5.1 & 5.2 3AB 4CDE 1DI 2E
10:30-17
Wed 25.08 3AB 6AB 1DII 4CDE
10:30-17 Demo Demo Demo Demo Demo
Thu 26.08 8DE 8DE 8DE 8DE 8DE
10:30-17
Fri 27.08 Student data presentation & Report writing
Sat 28.08 Report writing
Sun 29.08 Report writing
4CONTENTS
General introduction ................................................................................................................6
EXERCISE 1: Determination of sediment characteristics (density, water content, organic
content and chlorophyll a content) .................................................................11
EXERCISE 2: Measurement of sediment oxygen, carbon dioxide and dissolved
inorganic nitrogen exchange in light and darkness ........................................15
EXERCISE 3: Determination of O2 microdistribution and diffusive O2 exchange in
surface sediments ...........................................................................................20
EXERCISE 4: Measurement of sulfate reduction by the 35S technique .................................24
EXERCISE 5.1: Measurement of denitrification in the sediment by the 15N isotope-
pairing technique ............................................................................................29
EXERCISE 5.2: Measurement of potential nitrification (ammonium oxidation) in
sediment slurry. ..............................................................................................33
EXERCISE 6: Determination of vertical TCO2, DIN (NH4+ and NO2-+NO3-) and SO42-
profiles in the sediment ..................................................................................35
EXERCISE 7: Determination of particulate iron profiles in the sediment .............................37
EXERCISE 8: Molecular analysis of microbial community structure and function ..............40
Reference list ........................................................................................................................48
Appendix 1: Sediment collection ............................................................................................50
Appendix 2: Tables for exercise1,2,4,6,7…………………………………………………..51
Appendix 3: How to do tricky calculations.............................................................................62
Appendix 4: Safety regulations for laboratory work...............................................................64
Appendix 5: Manual for report writing ...................................................................................66
5Introduction
GENERAL INTRODUCTION
Organic matter enters coastal sediments either by sedimentation of phytoplankton and
terrestrial derived particles (sewage and rivers) or via primary production by benthic
microalgae (diatoms and cyanobacteria) and macrophytes (algae and seagrasses). Organic
particles located at the sediment surface will gradually be buried within the sediment by
continued sedimentation and faunal particle reworking (bioturbation). The rate by which the
burial occurs depends on the sedimentation/erosion regime and the density of benthic
macrofauna at any specific location. Organic matter in sediments is degraded (mineralized) by
an array of aerobic and anaerobic microbial processes with a concurrent release of inorganic
nutrients. The actual rates of decay depend primarily on organic matter quality (i.e., the
content of protein, cellulose, lignin etc.), age (decomposition stage) and temperature (season).
The chemical composition of organic matter in marine environments can be generalized by
the following formula:
(CH2O)x(NH3)y(H3PO4)z
where x, y and z vary depending on the origin and age of the material.
Decomposition in the upper part of the sediment occurs under aerobic conditions with
oxygen as electron acceptor by the following stoichiometry:
(CH2O)x(NH3)y(H3PO4)z + xO2 + yH2O → xHCO3- +yNH4+ + zPO43- + (x+3z)H+ + yOH-
As the oxic (oxygen containing) zone in coastal sediments usually is limited to the
upper 1-5 mm, a large fraction of the organic matter will be buried more or less
undecomposed into anoxic layers. In these layers the large and normally complex organic
molecules will be split into smaller and water-soluble units under the production of energy by
hydrolyzing and fermenting bacteria. These smaller units will then be degraded completely by
an array of microorganisms using a variety of oxidized inorganic compounds as electron
acceptors. These processes generally occur in the following sequence with depth in the
sediment: Mn4+ ≈ NO3-, Fe3+, SO42- and CO2 respiration (Fig. 1). The actual sequence is
determined by the ability of each electron acceptor to receive electrons, and thus the energy
output per degraded organic carbon atom.
Most of the major microbial processes are carried out by specific groups of organisms,
each of which is specialized in one or a few processes only. No single organism can catalyze
all the different processes. Oxygen-respiring bacteria may be facultative aerobes that grow by
denitrification or fermentation when oxygen is absent. Most manganese- and iron-reducing
bacteria, on the other hand, as well as all sulfate-reducing bacteria and methanogenic archaea
are strict anaerobes and are specialized in their use of electron acceptor. Furthermore, the
strict anaerobes cannot oxidize complex organic matter but rely on fermenting bacteria to
produce their substrates, of which acetate, other short-chain fatty acids, and hydrogen are the
most important. The sequence of anaerobic processes observed in the sediment (Fig. 1) is the
6Introduction
result of competition between the different respiratory groups for these common substrates.
The transition from one electron acceptor to the other downwards in the sediment will occur
when the most favorable is exhausted. ”When the best is gone, one has to accept something
less good.” Nevertheless, the usually observed decreasing degradation rate with depth in
sediments is not caused by the less efficient electron acceptors in the deeper layers, but rather
by decreasing quality of the organic matter (lability or degradability) with depth. However,
the variety of organic substrates which can be degraded seems to be narrower when moving
through the sequence of electron acceptors from O2 to CO2.
RESP Concentration (arbitrary scale)
Oxygen
O2 Mn
4+
Mangan. -
NO3
Nitrate
3+
Fe
Iron
Depth in sediment
2-
SO4
Sulfate
-
HCO3
Methanogen.
Figure 1. Idealized depth distribution of electron acceptors in marine sediment. The sequence of
respiration processes is indicated along the left vertical axis.
The quantitative significance of each of the various aerobic and anaerobic processes
for the total decomposition in any sediment is determined by several local conditions, such as
sedimentation rate, bioturbation rate, temperature etc. Oxygen respiration and sulfate
reduction are usually considered to contribute by up to 50% each. However, the latter process
can be responsible for up to almost 100% in organic-rich sediments. A process like
denitrification is usually of limited quantitative significance for mineralization (few percent)
due to low availability of NO3-. Although sulfate reduction is one of the least favorable
respiration processes, the high concentration of SO42- in seawater (300-1000 times higher than
7Introduction
O2) is responsible for its deep vertical distribution and thus quantitative importance. Inorganic
C, N and P compounds (HCO3-, NH4+ and PO43-), which are released by the mineralization
processes, will accumulate in the sediment porewater and gradually be transported upwards
by molecular diffusion (or bioturbation). Eventually they will all end up in the overlying
water column and once again act as substrate for primary producers (plants).
A large fraction of sediment oxygen uptake is not caused by aerobic respiration
(decomposition), but rather by reoxidation of reduced inorganic metabolites (e.g., H2S and
NH4+). About 50% or more of the sediment oxygen uptake is usually consumed by
reoxidation, which occurs when reduced compounds diffuse from deeper layers and up to the
oxic sediment surface. Reoxidation can be a pure chemical process, but usually it is mediated
by microorganisms. The oxidation of reduced compounds releases energy, which drives the
metabolism of chemoautotrophic Bacteria and Archaea. The most important of these
processes in sediments are nitrification and sulfide oxidation. Although the latter process is
quantitatively most important in terms of oxygen uptake, only the former process will be dealt
with here, since it is very important for nitrogen cycling in sediments. Nitrification is
generally a two-step process driven by aerobic Bacteria and Archaea. First NH4+ that is
released during ammonification of the organic matreial if oxidized to NO2- that subsequently
is oxidoixed to NO3-. The produced NO3- will diffuse either downwards into the sediment or
upwards to the overlying water column. If downwards, NO3- will typically be denitrified to N2
in anoxic layers and thus become unavailable for most living organisms. If upwards, NO3-
will be an essential nutrient for primary producers. About 30-50% of all NH4+ being produced
in sediments will normally be lost by coupled nitrification-denitrification. Despite its limited
influence on the carbon cycling, denitrification is very important for the nitrogen cycling in
sediments.
Until very recently it was difficult to obtain reliable information about the nature of
the microorganisms responsible for the chemical processes in sediments. Unlike animals and
plants, most bacteria and archaea cannot be identified simply by their size and shape. Only a
minority of the microorganisms present in a particular environment can be cultivated using
currently available techniques. This makes a comprehensive description of bacterial diversity
in the environment using classic microbiologic methods impossible. The solution to this
problem is the use of molecular sequence information to investigate microbial diversity and
identify prokaryotic species, an approach known as “molecular microbial ecology approach”.
The sequence of DNA bases in a particular region of the genome is characteristic of a
particular type of microbe and can be used for identification. The most commonly used genes
are those encoding for the ribosomal RNA molecules, which are structural components of the
ribosomes, particularly the 16S rRNA of the small subunit. This is certain to be present in all
bacterial cells whatever their type of energy metabolism or ecological niche.
The purpose of these laboratory exercises is to quantify processes driven by the most
important microorganisms and their diversity in 2 different coastal sediments. Sediment will
be obtained from a relatively organic-rich site, Kærby Fed (KF) in Odense Fjord, and from an
organic-poor site, Fællesstrand (FS) at Fyns Hoved. You will be split into 5 teams - team 1-3
will work with Kærby Fed sediment and team 3 and 4 with Fællesstrand sediment. During the
8Introduction
exercises, oxygen, carbon dioxide and dissolved inorganic nitrogen (DIN) exchange caused
by microalgal primary production and heterotrophic respiration in the sediment will be
measured (exercise 2). Subsequently, the small-scale dynamics of oxygen near the sediment-
water interface in light and darkness will be illustrated and related to microalgal biomass
(exercise 1 and 3). The rates of oxygen uptake and carbon dioxide release in the dark will be
related to rates of sulfate reduction (exercise 4) and denitrification (exercise 5A) in order to
determine the quantitative role of aerobic respiration, denitrification and sulfate reduction (see
Appendix 3). The distribution of oxidized and reduced particulate iron pools (Fe3+ and Fe2+)
will be examined (exercise 7) and compared to show the influence of processes related to
redox dynamics of iron. The oxidized iron pool will also be used to quantify the role of iron
reduction for the total sediment metabolism (see Appendix 3). Exercise 6 (porewater profiles
of TCO2, dissolved inorganic nitrogen and SO42-) and exercise 1 (porosity and organic
content) will contribute with essential data for the final calculation and interpretation of
results obtained in the other exercises. Potential nitrification will be determined in exercise 5B
and related to the results from exercise 8, where the distribution of marker genes for ammonia
oxidation will be quantified.
Exercise 8 illustrates how molecular techniques based on the analysis of
environmental DNA can be used to obtain information about microbial communities,
avoiding the need to culture the microbes. We will be using quantitative PCR (qPCR) to
determine the metabolic potential of the community for different processes in the nitrogen
cycle, namely nitrification and denitrification. By quantifying the copy numbers of the
functional marker genes for nitrification (ammonia monooxygenase; amoA gene) and
denitrification (nitrite reductase; nirS gene) in parallel to the 16S rRNA gene we will be able
to estimate what percentage of the community has the capability for performing these
processes. In order to be able to run this analysis, we will extract metagenomic DNA (also
called community DNA) from sediment samples. Here we will use a special method that
avoids the co-extraction of contaminating substances like humic acids, resulting in reasonably
pure metagenomic DNA usable for downstream molecular work. The marker genes will
subsequently be amplified by PCR and the amount of PCR product will be determined during
each amplification cycle using a double-stranded DNA binding fluorophore and a detection
system in the qPCR machine. Using this information will allow us to calculate back to the
copy numbers of the marker genes in the original sample and compare them with the potential
nitrification measurements from exercise 5,2 and the potential denitrification rates from
exercise 5,1.
You will find a precise time schedule for the work of each team during the various
exercises in the front of this manual (Appendix 1). The laboratory setup for the experimental
work has been done in advance, and each exercise has a specific location in the laboratory and
is indicated by its number. Please follow the instructions and avoid moving equipment around
in the laboratory without informing the instructors.
The sediment cores to be used during the course will be sampled by the students on
selected dates under guidance of the teachers. Make your own choice of when to participate in
this essential part of the course, but everyone must go at least one time. Please read the safety
9Introduction
instruction sheets (Appendix 4) – particularly the instructions on safe handling of
radioactivity before performing exercise 4.
All data obtained in exercise 1, 2, 4, 6 and 7 should be filled into the appropriate tables
found in Appendix 2. They must be sent by e-mail to the teachers when completed. At the
end, the results obtained by all teams will be available at the course web-site for your use
when writing the final report. Reports should be submitted as team-reports. All members of
the teams must participate on equal terms during laboratory work and result treatment as well
as during report planning, writing (guidelines for writing a report is given in Appendix 5) and
oral presentation during the final synthesis.
10Exercise 1
EXERCISE 1: Determination of sediment characteristics (density, water content,
organic content and chlorophyll a content)
Water content, density, organic content, and chlorophyll a content of sediments are important
supporting parameters to fully understand many aspects of microbial ecology in sedimentary
environments. When related to sediment density, the water content describes the volume of
pore spaces in the sediment (porosity, φ = volume water/total volume). This parameter is
usually used when microbial rates have to be transformed from per weight unit to per unit
volume of sediment. Raw data from laboratory experiments are, of practical reasons, usually
determined per unit weight, but they have a better ecological meaning when presented per unit
volume.
Organic content (loss on ignition) provides an estimate of the potential substrate for
decomposing bacteria. There is, however, rarely a simple relationship between organic
content and sediment metabolism. Sediment metabolism is more related to the quality and
degradability of the organic matter. Most of the organic matter determined as loss on ignition
is actually composed of refractory organic compounds of low degradability. It is important to
discriminate between the “organic matter content” and the “organic carbon content” which
sometime is mixed up in the literature.
Chlorophyll a content is a measure of microalgal biomass (diatoms and cyanobacteria)
and should be related to benthic primary production as measured in Exercise 2. The most
important input of labile organic matter at the two study locations is from benthic microalgae.
The biomass of benthic microalgae will here be quantified as chlorophyll a content in the
uppermost sediment layers. Chlorophyll a can, however, be relatively persistent to
degradation and pigments present in deeper layers are not expected to contribute to the
photosynthetic activity.
I. Materials:
2 sediment cores from the study sites (5 cm i.d.)
Slicing plates + stands + rulers + pistons
Alu-trays + alu-foil
4 cut-off syringes (5 ml)
8 centrifuge tubes (15 ml plast)
90% ethanol
2 M HCl
Plast spoons
Plast cuvettes
Pasteur pipets
Balance
Centrifuge
Spectrophotometer
Oven, trays from oven
16 crucibles
Muffle furnace.
Desiccator + separator plates
11Exercise 1
Vortex
Pencils
II. Procedures.
A. Slicing cores and determination of density
Slice two sediment cores, which has been taken with 5 cm i.d. core liners at the selected
locality, one at the time in the following intervals: 0-1, 1-2, 2-3, 3-4, 4-5, 5-6, 6-8, 8-10 cm.
Place the piston in the core liner (tight, to avoid loss of porewater) instead of the lower rubber
stopper (the upper stopper has to remain tightly in place). The core is then fixed on the stand
and the upper stopper is removed (Fig. 2). The first 6 slices are cut in 1 cm intervals and the
remaining 2 slices in 2 cm intervals by the use of the ruler and cutting plate. The slicing is
done by carefully pushing the sediment core inside the liner up to the chosen thickness as
indicated by the ruler. Then the cutting plate is pushed along the upper edge of the core liner
leaving the intact sediment slice on the cutting plate (use a finger to prevent the slice to slide
of during cutting). The sediment slice is then transferred into a pre-weighed and -labeled
(with a pencil) alu-tray, which is kept ready. Before the next slice is pushed up, the cutting
plate should be cleaned with a paper towel. Cut 4 slices at the time, and handle these as
described below - before cutting the next 4 slices. This will minimize evaporation, and thus
reduce errors in the water content results.
The density of the sediment is determined as the weight of a known volume. Fill a pre-
weighed and -labeled 5-ml cut-off (i.e., the tip is cut off) plastic syringe with sediment to the
4-ml mark (avoid any air in the syringe) and weigh it again. Density is calculated as follows:
d = m/4 (g cm-3), where m is the sediment weight (i.e., final weight minus syringe weight).
Weigh the alu-tray + the remaining sediment and place it in an oven at 105ºC.
Ruler
Cutting plate
Sediment
Core tube
Piston
12Exercise 1
Figure 2. Schematic drawing of set-up for core slicing
B. Water content
When the alu-trays have been in the oven for about 12 hours (the teachers will take them out),
they are transferred to a desiccator. Weigh the alu-trays with the dried sediment.
Water content (β) is calculated from wet weight (ww) and dry weight (dw) as follows
(remember to subtract the weight of the alu-tray):
β = [(ww - dw)/ww] ⋅ 100 (%)
Porosity (φ) is calculated according to: φ = β ⋅ d/100.
Transfer subsamples of the dried sediment to pre-weighed and -labeled crucibles (do
not fill them more than 2/3). Weigh the crucibles + sediment and placed it in a muffle furnace
at 520ºC.
C. Organic content
When the crucibles have been in the muffle furnace for 10 h (the teachers will take them out),
they are transferred to a desiccator. Weigh all crucibles + sediment and dump the sediment in
the waste bucket.
Organic content or loss on ignition (LOI) is calculated from dry weight (dw) and ash
weight (aw) as follows (remember to subtract the weight of the crucibles):
LOI = [(dw - aw)/dw] ⋅ 100 (%)
D. Chlorophyll a content
We use a rather crude spectrophotometric method, which is easy to perform and reliable. It
may not give the exact concentration of chlorophyll, but it is excellent for comparisons
between sites.
Slice two cores into the following depth intervals: 0-0.5, 0.5-1, 1-2 and 2-3 cm. Mix
and homogenize the slices. Transfer a subsample (ca. 0.5 g each) from each depth interval
into a test tube containing 5 ml of 90% ethanol, which is placed on a balance (remember to
zero the balance before adding the sediment) and weigh the sediment sample. Shake or
whirly-mix the test tubes for about 30 seconds. Extract the sediment in darkness (5°C) until
next day. Shake the test tubes regularly. When the extraction has finished, the test tubes are
centrifuged for 10 min at 3000 rpm. Measure the absorbance of the supernatant at 665 and
750 nm before and after acidification by one drop of 2 M HCl and mixing. The chlorophyll a
content is then calculated as follows (Castle et al., 2011):
Chlorophyll a (μg/g) = 29.6(665o – 665a) v/m
Where 665o is the absorbance at 665 nm subtracted by the absorbance at 750 nm before
13Exercise 1
acidification; 665a is the absorbance at 665 nm subtracted by the absorbance at 750 nm after
acidification; v is the volume of ethanol (5 ml); and m is the weight (g) of extracted sediment.
Plot the chlorophyll a concentration as a function of depth in the sediment. Compare
the concentrations between the two locations.
III. Questions and Figures, which should be considered in writing the report
1. Plot the porosity, the LOI and Chl a as a function of the sediment depth for both sites.
2. Compare the two sites and discuss potential differences.
3. Why is it important that there is no air in the syringe during measurement of density?
4. Why should samples always stay in a desiccator after drying or combustion?
5. Why are samples for organic content combusted at 520ºC?
6. Why do we measure absorbance at both 665 and 750 nm for chlorophyll analysis?
7. Give possible sources of errors.
14Exercise 2
EXERCISE 2: Measurement of sediment oxygen, carbon dioxide and dissolved
inorganic nitrogen exchange in light and darkness.
Sediment primary production and total heterotrophic metabolism is determined by measuring
oxygen and carbon dioxide exchange across the sediment-water interface. At the same time,
the exchange of dissolved inorganic nitrogen (DIN = NH4+, NO2- and NO3-) between the
sediment and the overlying water is quantified. DIN flux across the sediment-water interface
is a net measure of all nitrogen transformations occurring within the sediment.
I. Materials:
6 sediment cores from the study site (5 cm i.d.)
50 l seawater from the study site
1 tank with core holder and central magnet
Air pumps with tubing and air stones
1 greenhouse lamp
Alu-foil
6 magnet collars and 6 transparent lids with O2 patches
60 ml syringe + tubing
0.45 µm disposable syringe filters
Computerized O2 measuring set up
”Flow injection/diffusion cell“ analyzer.
2 mM HCO3- standard
Thermometer.
12 ml Exetainers (6)
Salinity-meter (conductivity).
20 ml scintillation vials (for nutrient samples) + vial holder.
Plast test tubes
Flow Injection Analyzer (Lachat).
Plast bags.
Light sensor
II. Procedures.
A. Sediment cores
Sediment cores have been taken with 5 cm i.d. core liners at the location and pre-incubated in
the storage tank at the selected temperature (15ºC) in 12 h light : 12 h dark cycles (light will
be kept off today until start of the incubation). ). 6 cores have been collected for flux
incubation with a sediment dept of 15-20 cm. Wash hands before or wear gloves when
handling cores in the incubation tanks. Describe the sediment of each core with respect to
colour zonation and any traces of animal activity (i.e. burrows of Nereis diversicolor). This
can be done before or after the incubation. Wrap 3 of the cores in alu-foil (dark cores),but
leave the top open until initial sampling has been completed. Submerge these cores in the core
holder around the magnetic stirring unit of the incubation tank together with the 3 unwrapped
cores (light cores) such, that the water surface is at least 2 cm above the upper edge of the
core liners. The light cores must be placed directly under the greenhouse lamp. Be sure that
15Exercise 2
there is continuous aeration in the tank. Measure and note water temperature, light level and
salinity (with conductivity detector).
B. Dark and light flux incubation
After turning the light on, initial (start) samples (20 ml) are taken from the water phase inside
each of the 6 cores while still submerged using a 60-ml syringe for analysis of CO2 and DIN
(Dissolved Inorganic Nitrogen). Fill a 12-ml Exetainer (carbon dioxide analysis, see later).
Fix a syringe filter to the syringe and filter 15 ml sample to a 20-ml scintillation vial (DIN
analysis, see later). Store the CO2 samples at 5ºC and freeze the DIN samples immediately.
After finishing the initial sampling, place stirring collars in the middle of the water of each
core and seal the cores air tight with a transparent lid holding a small patch of oxygen
sensitive phorphyrin-derivate fixed to the inside (see later for explanation) on top of the core
liner (do it under water) and stick the glass stopper in the hole in the lid and place the black
cover on the three dark cores. Note the start time. IMPORTANT: The entire procedure is
conducted for one core at a time, i.e., when the start sample has been taken from one core, it is
sealed before sampling is started on another core. Terminate the incubation for one core at a
time after 2-3 hours and take the final samples for CO2 and DIN as described for the initial
samples - BUT with the exception that sampling is done through the small hole in the lid.
Note the precise time at the end of incubation for each core. Measure and note temperature
and the height of the water phase in the cores.
C. Quantifying abundance and biomass of Nereis diversicolor
The respiration contribution to O2 uptake and CO2 production by the most common burrowing
benthic fauna species, Nereis diversicolor, will be estimated from the abundance and biomass
in flux cores. All cores are therefore sieved through a 1 mm mesh to recover the worms.
Recovered worms are transferred to petri dishes (one per core). Worms are counted and
weighed. The weighing is done by blotting worms briefly on a paper tissue to remove excess
water before transferring them one by one to a petri dish containing seawater, which is placed
on a balance.
Respiration (V, µmol h-1) as a function of individual biomass (M, g wet wt.) for N.
diversicolor (15°C) is:
VO2 = 2.15 M0.68
Use the average individual biomass (M) and multiply by the density (m-2) of N. diversicolor
to get the contribution on an area basis.
D. Photochemical oxygen determination
Oxygen will be measured by the optode approach. Central for the technique is the ability of
oxygen to quench the fluorescence of certain chemical compounds. In other words, O2 can
absorb the energy that otherwise would have been sent out as fluorescence from a given
fluorophore. There exist numerous configurations, chemistries and procedures to quantify O2
availability by this approach (e.g. Klimant et al. 1997, Kuhl & Polerecky 2008) and many are
16Exercise 2
custom build and optimized for specific experimental approaches and procedures – some of
these will be demonstrated during the course. The system applied for flux measurements is a
commercial available system that is sold by the company Pyroscience. The system is called
Firesting and has 4 channels that can be applied for simultaneous measurements.
Small patches of an immobilized phorphyrin-derivate are fixed to the inside of the lids
to the sediment cores. This compound is excited by red light and the O2 sensitive emitted light
has an optimum in the near infrared region. The excitation light is supplied to the sensor patch
via a fiber placed in the small hole of the lids. The same fiber also guides the O2 sensitive
light back to the measuring instrument. The sensors are pre-calibrated and the O2 signal (in %
air saturation) over time during the incubation is directly read out on the computer screen and
can be noted and/or saved as a text file. Measure temperature and salinity for conversion from
% air saturation to concentration in µM (a small table will be provided ).
E. CO2 analysis (Hall & Aller 1992)
TCO2 will be analyzed on a ”flow injection/diffusion cell“ analyzer. Samples (0.1 ml)
containing CO2 are injected into a carrier stream of 45 mM HCl which is pumped into one
side of a diffusion cell equipped with a gas permeable membrane (plumbers tape). A receiver
stream of 10 mM NaOH is pumped simultaneously to the other side of the membrane. All
CO2 from the acid carrier side diffuses into the base receiver side. The amount of CO2 is then
detected as a reduction in conductivity (by a conductivity detector) as a result of base
neutralization in the receiver stream. The signal is recorded and peak height measured by a
computer.
Signals will be evaluated by the program Clarity go to “Analysis” and choose “set zero”, then
chose “run single” which activates the data collection. The time range (0-20 min) and signal
(-1 -10mV) can if needed be changed during a run. When the baseline is stable you are ready
to inject standards and samples. For the samples use a 5 ml syringe with a small piece of
tubing to take samples from the exetainers. With the valve in the “load” position inject l ml
into the sampling port to fill the 0.1 ml sample loop. Now turn the injection port to the
”inject“ position. Leave the syringe in the sampling port. The CO2 peak appears within 1 min,
and when the baseline is stable again, inject sample once more by turning the injection port
back to “load” and “inject” positions, repeat 3-4 times. Stop run by the stop icon (menu),
which opens the data collection site. Check if baseline is okay or change it manually, note
peak height. Close date collection in the upper right corner and start the next injections. The
concentration of TCO2 is calculated manually based on the 2mM standard. After
measurements flush the system with Milli-Q for 15min (place tubing in Milli-Q), stop pump
and open clamps, turn off conductivity meter, empty waste containers), refill HCl and NaOH
bottles and shut down the computer
F. DIN analysis
Analysis of NH4+, NO2- + NO3- is done spectrophotometrically on a LACHAT Flow Injection
Analyzer (the exact procedure will be explained during the exercise). NH4+ is analyzed
according to the Berthelot-reaction (Bower & Holm-Hansen 1980). In a weak basic solution
(pH 9.5-11), NH3 reacts with hypochlorite to produce monochloramine, which in turn
17Exercise 2
provides indophenol blue in the presence of salicylate, nitroprusside-ions (catalyst) and
surplus of hypochlorite. The automated NO2- analysis is based on a reaction between the NO2-
ion and sulfanilamide under acid conditions. This produces a diazo-compound which is
coupled with N-(1-naphthyl)-ethylene-diamine to provide a purple azo-dye (Armstrong et al.
1967). The absorption of the color reaction is determined at 550 nm. The analysis of NO3- is
based on a reduction of NO3- to NO2- by a copper-cadmium column followed by the analysis
as described above for NO2-. Procedure: Transfer samples (in the appropriate dilution) to the
small autosampler cups and place them in the autosampler. From here, they automatically will
be mixed with reagents and measured in a spectrophotometer one by one. The concentrations
(in μM) are calculated from a relationship based on the peak areas of 5 known standards
(NH4+: 0, 10, 20, 30, 40 μM; NO3-: 0, 5.0, 10.0, 20.0 μM). NO2- and NO3- will not be
determined separately here because the concentration of NO2- is very low. Results should
therefore be presented as NO2- + NO3-
Calculation of fluxes: Oxygen exchange per m2 sediment surface per day (JO2, mmol
m-2 d-1) is determined from the slope (α) of a linear regression of O2 against time (days)
according to:
Flux = slope ⋅ water volume / surface area
JO2 = α ⋅ π r2 h / π r2
-2 -1
mmol m d = mmol m-3 d-1 ⋅ m3 / m2
This can be reduced to: JO2 = α ⋅ h where the slope α has the units of μM d-1 (μmol
dm-3 d-1 = mmol m-3 d-1) and h is the height of the water column above the sediment in m (or
cm/100)
Carbon dioxide (JCO2) and DIN exchange (JDIN) per m2 sediment surface per day
(mmol m-2 d-1) are calculated according to:
Flux = change in concentration ⋅ water volume / surface area / time
Jx = (Ce – Cs) ⋅ π r2 h / π r2 / t (values)
-2 -1 -3 3 2
mmol m d = mmol m ⋅ m / m / d (units)
This can be reduced to: Jx = (Ce – Cs) ⋅ h / t where Ce and Cs are end and start
concentration in μM (μmol dm-3 = mmol m-3), h is the height of the water column above the
sediment in m (or cm/100) and t is incubation time in days (or hours/24).
III. Questions & Figures, which should be considered in the report
1. Provide a Table and a diagram of the total exchange rates of O2, DIN and TCO2 for
the two sites.
2. Calculate RQ (respiratory quotient) and PQ (production quotient) from the two sites
and explain what this tells us about the two sites (see also appendix 3)?
3. What is the contribution of Nereis respiration to the total O2 uptake (TOU) measured
in darkness at the two sites.
18Exercise 2
4. Calculate the CO2/DIN flux ratio for the two sites and explain what it tells us?
5. Which processes within the sediment determine the net flux of DIN?
6. Why is it necessary with stirring during flux incubations?
7. What can be the cause for observed differences in exchange rates among cores from
the same site?
8. Give possible sources of errors.
19Exercise 3
EXERCISE 3: Determination of O2 microdistribution and diffusive O2 exchange in
surface sediments
Photic surface sediments are characterized by very steep O2 gradients reflecting concurrent
production and consumption of O2. Fast microsensors are required to resolve the distribution
and the temporal dynamic of O2 in such environments – in this exercise we use Clark-type O2
microelectrodes (Revsbech & Ward 1983).
The microprofile approach makes it possible to quantify O2 exchange and penetration
in darkness and light, in order to calculate the O2 consumption in darkness and the net
photosynthesis in light. Give some assumption it is in principle possible to calculate the gross
primary production from such pared datasets. It is, however, also possible to measure the
gross photosynthesis directly by the so-called light-dark shift technique (Glud et al. 1992), but
that will not be done in the current exercise.
I. Materials:
1 sediment core
Complete automated microelectrode measuring set up
Adjustable light source
2 stands with hooks
Light logger
Diagram for diurnal down-welling irradiance
Air-pump with tubing and air-stone
Software for O2 solubility
Table for diffusion coefficients
Flow-through chamber
Aquaria pump
II. Procedures:
A. Dark O2 penetration and diffusive mediated exchange:
Experimental: The microelectrode tip is placed 1-2 mm above the sediment surface.
Subsequently the sensor is lowered in steps of 50 µm (0.05 mm), while the O2 concentration
at each depth is recorded and stored by the computerized set-up. Note when the sensor enters
the sediment. Repeat the measurements until you record low stable signals (i.e 0-3 mV) that
remain constant when you move the sensor - this is defined as the anoxic zone. The sensor is
moved back to the start position, moved 3-5 mm horizontally and the measuring procedure is
repeated. You should measure a minimum of three profiles – but are welcome to measure
more.
Calculations and theoretical work: The recorded text-files are transferred into Excel and here
the first step is to calibrate the recorded signals into µmol L-1. Oxygen sensors have a linear
20Exercise 3
response and as we know the O2 concentration in the overlying water phase at the given
temperature and salinity and the signal size in the anoxic (zero oxygen) sediment layers, it is
simple to calibrate each of the recorded data points according to:
Calibrated signal (µmol L-1) = Sr (Cw /(Sw-Sa)) + Sa(Cw /(Sw-Sa))
Where Sr is the recorded signal (in mV), Sw the signal in the overlying water phase, Sa the
signal in anoxic sediment and Cw is the O2 concentration in the overlying water phase. Define
the relative position of the sediment surface. Assign this position to depth “0” and use
negative prefix for recordings at any depths below this fixed point (see Fig 3). Plot the
respective microprofiles with “Depth” (mm) on the ordinate and O2 concentration µmol L-1)
on the abscissa.
Determine the average O2 penetration (P) (unit: mm) and calculate the average
Diffusive O2 Exchange (DOE – Jdark,up) from the profiles using:
DOE (mmol m-2 d-1) = -Do (dC/dZ)*8640
Where Do is the molecular diffusion coefficient (cm2 s-1) at the given conditions (see Table 1)
and (dC/dZ) is the slope of the linear O2 concentration gradient in the Diffusive Boundary
Layer (DBL) (µmol L-1 mm-1).
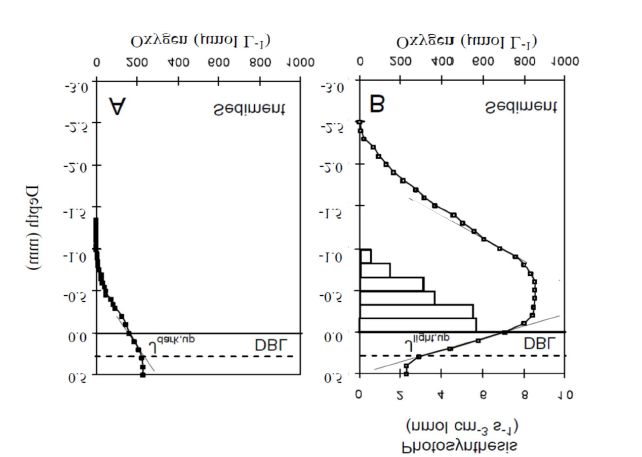
Figure 3. Two microprofiles measured in sediment cores recovered from a water
depth of 1.2 m in Helsingør Harbour, Denmark. The profiles were measured at the
exact same spot in darkness and at a irradiance of 600 µmol photons m-2 s-1. The
white horizontal bars indicate the gross photosynthetic rates at each depth measured
by the light-dark-shift approach (not measured in this exercise). The temperature was
1 ºC and the salinity 12.
21Exercise 3
B. O2 penetration and diffusive mediated exchange as a function of light:
The experimental procedure in II.A is repeated three times at 4 different irradiances (i.e. 10,
50, 100. 300, µmol photons m-2 s-1). Start with the lowest irradiance. Calibrate and plot the
respective profiles as above. Determine the average O2 penetration (P) (mm) at the respective
light levels.
Calculate net photosynthesis (Net-P, mmol m-2 d-1) in the photic zone for each light
level using a modified version of the DOE equation above (see also Fig 3B):
Net-P = DOE (Jlight,up) = ((Do (dC/dZ)) *8640
III. Questions and figures to be considered in the report:
1. Present one set of profiles including the one measured in darkness and at the four
respective light levels for both sites. See Fig 3 for units and layout.
2. Describe and discuss the shape of the profiles.
3. Present the derived O2 penetration depth and the DOE/NetPP as a function of the light
intensity for the compiled datasets from the two respective sites. What can be learned
from such a figure?
4. How does DOE measured in darkness compare to the total oxygen uptake (TOU)
measured during intact core incubations? Discuss potential reasons for any
differences.
22Exercise 3
5. Is the sediment net heterotrophic or autotrophic when integrated over a normal 24h
day/light cycle?
6. Discuss if the resolved benthic primary production is important on system level.
23Exercise 4
EXERCISE 4: Measurement of sulfate reduction by the 35S technique
The purpose of this exercise is to determine the rate of sulfate reduction in the sediment.
Reduction of sulfate to sulfide is followed by the use of the radioactive isotope 35S. Labeled
sulfate in the form of 35S-SO42- is injected into the sediment (Jørgensen 1978). The rate of
sulfate reduction can be determined when the ratio between the added amount of labeled
sulfate and the amount of sulfur recovered as labeled reduced sulfur compounds is related to
the total concentration of sulfate in the sediment.
Radioactive sulfide is incorporated into various reduced sulfur pools in sediments. In order to
release these pools, we will distill the sediment according to the TRIS technique (Total
Reduced Inorganic Sulfur). By this technique, both the acid volatile sulfides and the
chromium reducible sulfur will be released simultaneously (Fossing & Jørgensen 1989). Acid
volatile sulfides (FeS) are converted to H2S by the addition of hydrochloric acid (HCl). To
release the chromium reducible sulfur (FeS2 and S0) in the form of H2S, on the other hand, a
reducing agent (chromium II) is needed. The following process describes pyrite (FeS2)
reduction: FeS2 + 2 Cr2+ + 4 H+ → Fe2+ + 2 H2S + 2 Cr3+
I. Materials:
2 sediment cores in tubes with 1 injection port per cm. (2.6 cm i.d.)
Stand + holder for tubes
Cutting plate + ruler + piston
2x6 plastic centrifuge tubes (50 ml)
Centrifuge
Balance
Whirli mixer
Spectrophotometer
Cuvettes (1 cm)
Gloves
Scintillation counter
2 x 6 reaction flasks + various for distillation
Beaker for dist. H2O.
Long test tubes
20 ml scintillation vials.
7 ml scintillation vials.
Vial holders
10 ml plast centrifuge tubes
Eppendorf tubes
Waste container
Plastic sheet for covering table
60 ml syringe
5 ml Finnpipette + tips
1 ml Finnpipette + tips
24Exercise 4
200 μl Finnpipette + tips
50 μl Hamilton syringe
Long and short pasteur pipettes
Radiotracer, 35S-SO42-, carrier free 12 kBq µl-1
20 % og 5% (w/v) Zinc acetate.
6 M HCl
Chromium solution (1 M Cr2+ in 2 M HCl).
Scintillation liquid
Cline’s reagent (“low Cline”).
Silicone oil (anti foam)
II. Procedures (This exercise is done in a marked and approved area. Only persons
participating in the exercise are allowed in this area. Lab coats will be handed out and gloves
must be worn)
Fill out Table 8 in appendix 2 as you progress!
A. Incubation (Before you start read the radio-safety instruction sheet - Appendix 3)
Two sediment cores have been sampled using core liners (2.6 cm i.d.) equipped with silicone
sealed injection ports for every cm. Mount the cores on the lab-stand and remove the
overlying water. Inject 5 μl of a 35S-SO42- solution (60 kBq) in every injection port down to
10 cm depth by leaving the tracer as a horizontal line while pulling the syringe and needle
backwards. Allow the cores to incubate for 4-5 hours in darkness at the same temperature as
the flux cores (Exercise 2). Note the time of start and temperature.
B. Core sectioning
Label and pre-weigh (with lids) 2x6 plastic centrifuge tubes (50-ml). Add 5.0 ml (1 cm slices)
or 10.0 ml (2 cm slices) of 20% ZnAc (density 1.03 g/ml) and re-weigh the tubes. Mount the
incubated cores on the lab-stand. Cut 6 sections (0-1, 1-2, 2-4, 4-6, 6-8, 8-10 cm) as described
in Exercise 1 and transfer them to the centrifuge tubes. Clean the cutting plate with paper
towels between each slice. Cap the tubes immediately and mix the content by shaking before
weighing the tubes again. Note the time of sectioning. Finally, all samples are stored frozen.
Transfer the remaining sediment to the plastic bag labeled radioactive waste.
C. Distillation
Thaw the frozen samples in the centrifuge tubes and centrifuge them at 3000 rpm for 10
minutes. Transfer 200 μl of the supernatant into a 7-ml scintillation vial (label it on the lid!)
for determination of 35S-SO42- activity, and then add 800 μl of distilled water. Scintillation
liquid will be added later (see below). Transfer an additional 1 ml supernatant to 1.5 ml
Eppendorf tube (a spare sample in case of problems with the original) before decanting the
remaining supernatant to the waste container. Weigh the centrifuge tubes with sediment again
after removing the supernatant.
25Exercise 4
While the thawing proceeds, the ZnAc traps for retaining H2S are prepared (H2S is
precipitated as ZnS). Add 10 ml of 5% ZnAc into 6 long test tubes together with one very
small drop of silicone oil (prevents foaming). Mount the traps in the distillation unit.
Mix the sediment in the plastic centrifuge tubes (after centrifugation and decanting the
supernatant) and transfer and weigh about 4 g of sediment (msub) into a reaction flask.
Transfer then 15 ml of distilled water into each reaction flask and seal all by stoppers. When
all reaction flasks are placed on the heating plates, the entire distillation unit is assembled by
sealing the ”tops” equipped with long Pasteur pipettes. Turn on the cooling water!!!
Remember to check the water flow. Turn on the N2 flow using the small valves above the
distillation unit. The flow is appropriate when there is a calm and steady bubble development
in the traps. Let the reaction flasks stand with bubbling for 5-10 minutes before adding 8 ml
of 6 M HCl (use a plastic syringe) and 16 ml of a chromium solution (1 M Cr2+ into 0.5 M
HCl).
Turn on the heating plates (level 3). Let the distillation proceed for 45 minutes after
boil. Keep an eye on the N2 bubble pattern and the flow of cooling water. The reaction
mixture has to boil vigorously.
Terminate the distillation by turning off the heating and dismantling the unit
(disconnect the connection to the trap) and close the N2 flow. resuspend the content of the
traps with an automatic pipet and transfer immediately 5 ml into a 20-ml scintillation vial
(only labels on the lid). The remainder is transferred into 20-ml scintillation vials and stored
for later use.
Add 2 ml scintillation liquid to all 7-ml vials containing supernatant and 10 ml
scintillation liquid to the 20-ml vials containing trap material, and mix the solution for about 1
min. Make blanks. The samples are now ready for counting. Ask the teachers about the
procedure.
The reaction mixture from the reaction flasks is transferred into a waste container. The
reaction flasks are rinsed thoroughly with distilled water before the start of a new distillation.
Warning. Bottles with N2 are handled by staff members only. Working with open
samples must occur on the covered tables. All handling of open samples must be done
standing up, partly to avoid inhalation of aerosols, partly to minimize the risk of person
contamination. Never use a whirli-mixer for samples in open containers, instead you must mix
with an automatic pipette. 35S might be mutagenic. Waste must be collected in a plastic
container marked “waste (affald in Danish)”. A solution of 6 M HCl is very corrosive.
Vapours are very easily absorbed in the mouth, throat and lungs, where it can cause corrosion
and fluid diffusion (dys-pnoea, difficulty in breathing). Chrome chloride may be mutagenic.
Waste is collected in a plastic container marked “Cr”. Scintillation fluid is a mixture of
organic solvents, and inhalation may cause brain damage. Use the fume hood! All other
waste must be collected in the buckets with radioactive marks.
D. Calculation of sulfate reduction rate
The rate of sulfate reduction (SRR) is calculated according to:
26Exercise 4
[SO42-]s ⋅ a ⋅ 24 ⋅ 1.06
SRR = ────────────── (nmol SO42- cm-3 d-1)
(A + a) ⋅ h
where [SO42-]s is sulfate concentration in nmol cm-3 sediment = [SO42-] (mM) ⋅ φ ⋅ 1000
(determined from porewater profiles, see Exercise 6); a is the total radioactivity of sulfide
(H235S) in the sample slice (remember that only a subsample of the traps has been counted); A
is the total radioactivity of sulfate (35SO42-) in the sample slice; h is incubation time in hours;
1.06 is a correction factor for bacterial isotopic fractionation. Note that A and a have to be
transformed to dpm cm-3, based on the sediment weight and porosity (from Exercise 1) and
corrected for the amount of sample used in the distillation.
Calculation of a:
K ⋅ (dpma - dpmB) ⋅ msedc ⋅ d
a = ──────────────────── (dpm cm-3)
msub ⋅ msed
where: K = correction factor to scale to radioactivity in total trap volume (you may have
only counted the radioactivity of a part of this volume).
msed = weight of sediment slice before centrifugation (g).
msedc = weight of sediment after centrifugation and decantation of ZnAc (g).
msub = weight of sediment subsample (g).
dpma = radioactivity of H235S (dpm).
dpmB = background radioactivity (dpm).
d = sediment density (g cm-3). - from Exercise 1
Calculation of A:
(VZnAc + msed ⋅ ß/100) ⋅ (dpmA - dpmB) ⋅ d
A = ──────────────────────────── (dpm cm-3)
VA ⋅ msed
where: VZnAc = volume of ZnAc (5.0 or 10.0 ml).
ß = water content (%). - from Exercise 1
dpmA = radioactivity of 35SO42- (dpm).
VA = volume of sampled supernatant (0.2 ml).
E. Determination of the reduced sulfide pool
Besides the labeled sulfide, the distillation procedure has also released and trapped the total
pool of reduced inorganic sulfur in the sediment. The size of this pool can be determined by a
spectrophotometric technique. For this purpose, we will use the excess trap material, which
was stored in 20-ml scintillation vials. Mix the trap material thoroughly on a Whirli-mixer.
Transfer 50 µl (KF) or 200 μl (FS) of each sample (+ a blank consisting of 5% ZnAc) into
27Exercise 4
centrifuge tubes and dilute with 10 ml of distilled water. Add 800 μl of Cline’s reagent (Cline
1969) to the centrifuge tubes, close lids immediately, shake the tubes and let them rest for 20
minutes. Transfer ca. 3 ml of each sample to a cuvette and read the absorption at 670 nm on a
standard spectrophotometer. The absorbance must be below 0.8. If it is higher, the entire
procedure is repeated using less trap material.
Warning. Cline reagent contains 6 M HCl and is very corrosive. Take good care and
use paper cover on the table. The reagent also contains dimethylphenylendiamine (0.4%),
which is very easily absorbed through the skin and methaemoglobin is formed in the blood.
Dimethylphenylendiamine may have mutagenic and other dangerous effects (not yet
completely known). By reaction with sulfide, methylene blue is created, which can be
mutagenic. All Cline-work must be done in the fume hood. Gloves and lab coat must be
worn and waste must be collected in a plastic container marked “Cline”.
The concentration is calculated using a constant factor (μM/abs) based on the
absorption coefficient for the actual Cline reagent used here (will be handed out by the
teachers). The pool of Total Reduced Inorganic Sulfur (TRIS) can be obtained according to:
abss ⋅ F ⋅ b ⋅ msedc ⋅ d
[TRIS] = ──────────────── (μmol cm-3)
k ⋅ 1000 ⋅ msub ⋅ msed
where: abss = absorbance of trap sample.
k = absorption coefficient (abs/μM).
F = dilution factor.
b = trap volume (10 ml).
III. Questions and figures to be considered in the report
1. Plot SRR as a function of depth for both sites.
2. Plot TRIS as a function of depth in the sediment for both sites.
3. What are the advantages and disadvantages by incubations using intact cores?
4. Discuss the depth distribution of sulfate reduction rates in the sediment.
5. Compare the profiles of sulfate reduction rates with those of TRIS.
6. Calculated the depth-integrated sulfate reduction (mmol m-2 d-1).
7. Compare the depth-integrated SRR with sediment oxygen uptake (Exercise 2) - How
much of the oxygen may be used to oxidize H2S at the two sites? Discuss potential
differences.
8. How important is the SRR for the total benthic mineralization at both sites (see also
appendix 3)? Discuss potential differences.
9. Give possible sources of errors.
28Exercise 5
EXERCISE 5.1: Measurement of denitrification in the sediment by the 15N isotope-
pairing technique
Sediments typically house an active and extremely interesting microbially driven nitrogen
cycle. Nitrate is used in denitrification (as well as anammox and nitrate reduction to
ammonium) and this process can sometimes account for a significant fraction of carbon
mineralization in sediments while also representing an important sink for bioavailable
nitrogen. Denitrifiers are facultative anaerobic bacteria which can use nitrate instead of
oxygen in anoxic environments. Nitrate comes either from diffusion from the overlying water
or from nitrification in the sediments, a chemoautotrophic process where ammonium,
liberated to the sediment through organic matter decomposition, is oxidized nitrate. This
exercise will focus on denitrification.
Our determination of denitrification rates applies the Isotope Pairing Technique (IPT)
based on addition of nitrate labeled with the stable isotope 15N and subsequent analysis of the
labeled dinitrogen pairs formed (Nielsen 1992). Denitrification rates can in theory be
determined from the accumulation of N2 in the water column. In reality, however, this is
extremely difficult due to the high background concentrations (about 500 μM) of N2 in water.
Given some assumptions, the source of nitrate (overlying water or nitrification) for the
denitrification can be derived from the Isotope Pairing Technique. The IPT as applied here
assumes that anammox is not active. This assumption typically holds for shallow coastal
sediments.
I. Materials
3 sediment cores with sediment to about 10 cm from the top of the core
10 mM stock 15NO3- solution.
Alu foil and saturated ZnCl
Stirring collars
Sediment stirring set up similar to exercise 2
Plastic disposable pipets
Ruler
Glass spatulas
50 ml glass syringe
7 cm isoversinic (gastight) tube to fit syringe
10 ml Exetainers
10 ml plastic vials for nutrient samples
0.45 µm syringe filters
10 ml syringes
1 ml pipet and tips
200 µl pipet and tips
tape
markers
calculator
29Exercise 5
rack
II. Procedures
A. Incubation
Three sediment cores from your location are taken and the water level is adjusted to about 0.3
cm from the rim. Use a 50-ml syringe with a piece of tubing and try to avoid resuspension of
sediment. Then take a water sample for nutrient analysis from the circulating water in the tank
and filter it through 0.45 µm filters into a plastic vial. This and subsequent nutrient samples
are needed to determine the percentage of labeled nitrate for separation of the sources of
nitrate for denitrification (Dw and Dn – see below).
Before addition of 15NO3- the height of the water column is measured to calculate the
volume in which the added nitrate is diluted. Calculate the volume of your overlying water
and add enough stock solution to bring the 15NO3- concentration to 50 μM. Check this
calculation with the teachers before adding the 15NO3-. Mix 15NO3- into the water phase up by
gentle bubbling with air from a disposable pipette. Notice: 15N is not a radioisotope! Take a
subsample for NO3- analysis from each core and filter it into a plastic vial. Remember to label
the vials with team number, core number and time (before or after label addition).
Note starting time, add a stirring magnet, and close the core with a stopper without
trapping air bubbles. Do not press the stopper too hard. Leave the cores with circulation in
darkness for about 3 hours.
At the end of the incubation, note the time and stop denitrification by dripping ~0.5 ml
saturated ZnCl2 solution (a little toxic and corrosive) into the overlying water of the core with
a small plastic pipet. With a glass spatula, mix the top 6 cm of sediment and water with gentle
vertical and rotating motions to distribute the labeled N2 evenly before sampling. Avoid
skimming the surface as gas may escape. Check the new height of the water column and the
height of the mixed sediment to determine the volumes in which labeled dinitrogen will be
distributed. With a 50 ml syringe with a small piece of isoversinic tubing at the end, first take
in a few ml of the suspension and discharge to wash out any gas phase in the syringe, Now,
carefully withdraw about 30 ml sample. You will have prepared 6 Exetainers by adding two
drops of ZnCl2 solution to each. Transfer the sample to an Exetainer by introducing the tube
at the bottom and then draw up as the vial is filled to the rim. This is to minimize loss of
dinitrogen to the air. Put on the screw cap and check that no bubbles are trapped. Take two
samples from each core. Dinitrogen samples are marked with Team number and core number,
location and incubation time. They are saved for later IRMS analysis.
B. Analysis of dinitrogen by mass spectrometry (IRMS).
The isotopic composition in the produced N2 is determined by isotope ratio mass
spectroscopy. Initially, with the lid still on the Exetainers, one cm of sample is replaced with 2
ml helium through hypodermic needles inserted through the rubber seal, and by vigorous
shaking, dinitrogen is equilibrated into the He headspace. From the headspace, 0.25 ml is
30You can also read

















































