Amyotrophic Lateral Sclerosis (ALS) - aka Lou Gehrig's Disease presentation by Nat Royer
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

Amyotrophic Lateral Sclerosis (ALS)
aka
Lou Gehrig's Disease
presentation by
Nat Royer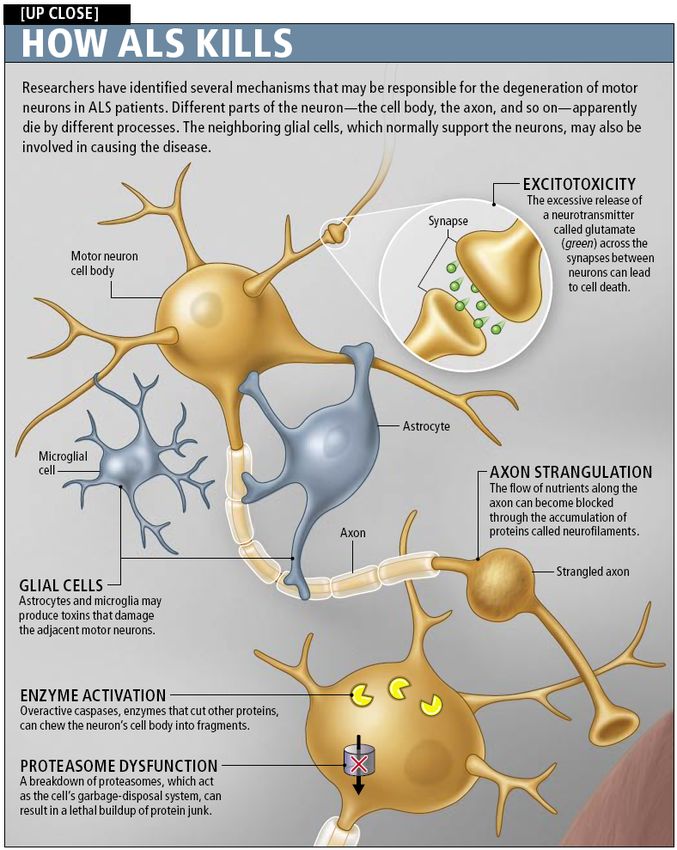
A (negative) myo (muscle) trophic (nourishment)
Lateral (location in spinal cord)
Sclerosis (hardening)
http://www.alsa.org/images/cms/Research/Topics/disease_process.jpg
Epidemiology
An estimated 30,000 Americans have ALS
Most are between the ages of 40-70
50% live more than 3 years, 10% live 10+
Two main types of ALS
− 90-95% of cases are sporadic ALS (SALS)
− 5-10% have familial ALS (FALS)
− Guamanian
Notable People with ALS
http://www.medinah11.net/bohlso http://www.depletedcranium.com/h http://images1.wikia.nocookie.net/m
n/Lou_Gehrig_files/image001.jpg appyscience/Stephen_Hawking.jp uppet/images/7/78/Writer.jonstone.jp
g g
Lou Gehrig Stephen Jon Stone
Hawkings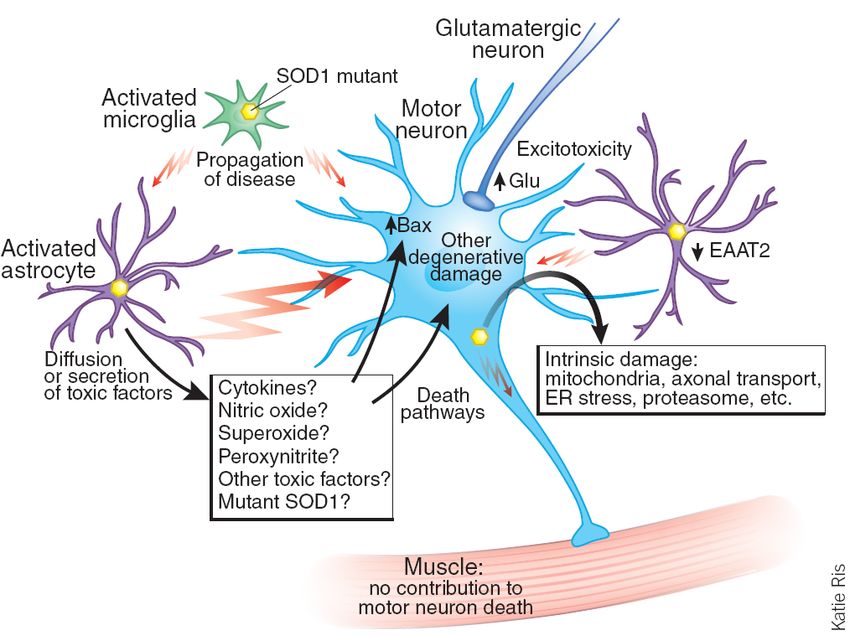
Signs & Symptoms
Early symptoms: muscle weakness,
tripping, slurred speech, muscle
twitches
Defective upper motor neurons:
− Stiff muscles and exaggerated
reflexes
Defective lower motor neurons:
− Muscle weakness, cramps, and
twitches
Eventual trouble swallowing,
breathing, and standing, but usually
retain all cognitive ability
Familial ALS
10 year later age of onset
than SALS
Rarely exhibits typical
dominant or recessive
inheritance
A few examples of
complete penetrance in
families have been
studied to identify mutated
genes
SOD1 in FALS
In 1993 researchers discovered SOD1
mutations in patients showing autosomal-
dominant FALS
Seen in 20% of FALS patients
Over 100 different mutations in SOD1 have
been shown to lead to ALS
mSOD1 is often polyubiquitinated and forms
aggregates
SOD1 in FALS

What makes mSOD1 neurotoxic?
mSOD1 seems to cause ALS without affecting
enzymatic activity
May increase the unfolded states of SOD1 and
lead to protein aggregates
Could cause malfunction of proteasomal
systems
Might interact with dynein and interfere with
retrograde axonal transport
mSOD1 and ER stress
Suppression of
protein translation
Induction of ER
chaperones
Degradation of
misfolded proteins via
the ubiquitin-
proteasome pathway
Induction of
apoptosisGlial cell damage can contribute to ALS
Jean-Pierre Julien found that astrocytes with
SOD1 mutation can exacerbate neuronal death
Removing mSOD1 from non-neuronal cells
slows the progression of the disease
Nagai et al. showed that this destructive
relationship is specific between astrocytes and
motor neurons
The toxic factors released by astrocytes are still
undeterminedGlial cell damage can contribute to ALS
Glutamate excitotoxicity and ALS
Spreux-Varoquaux et al. found that 40% of 400
patients with sporadic ALS had increased
glutamate levels in cerebrospinal fluid
Astrocytic glutamate transporter EAAT2 is
responsible for clearance of synaptic glutamate
Mutations in SOD1 have been shown to
severely reduce levels of spinal EAAT2
Repetitive firing leads to neuronal deathGlutamate excitotoxicity and ALS
Neurofilaments and ALS
Many ALS patients display accumulation of
neurofilaments in cell body and proximal axons
Studies have shown that increasing
neurofilaments in perikarya and decreasing
them in axons is protective against ALS
Perikarya accumulation could serve as a buffer
against excessive Ca2+ levels and
hyperphosphorylation of neuronal substrates
Axonal accumulation interferes with axonal
transportOther possible causes of ALS
Chronic activation of caspases causing
apoptosis
Deletion of hypoxia-response element in the
VEGF gene
Autoimmune response against motor neurons
What can we do for therapies?Treatments – NAD synthesis
The majority of ALS cases have
an unknown cause, so the goal
is to treat neuron damage
Jeffery Milbrandt at WashU
demonstrated that mice with
increased ability to synthesize
NAD had slowed axon
degeneration when injured
Resveratrol is a small molecule
that can activate NAD production
and cross blood-brain barrierTreatment with growth factors
Defective VEGF has been shown to lead to
neuron degeneration
Treatment with VEGF can delay onset and slow
progression of ALS in mice
IGF-1 has also been successful in extending life
span of mice with motor neuron disease
How do we get these larger molecules into the
central nervous system?Other treatments
Stems cells - can migrate to site of damaged
neurons and release important growth factors
RNAi - can slow down ALS by shutting down
defective SOD1 using RNAi
Exercise - can stimulate new neuron growth
and increase levels of growth factors
Drugs - Riluzole is first FDA approved drug
treatment of ALS and works by decreasing the
release of glutamateThe future of ALS research
Various studies have reported gene mutations
of TDP-43 in both rare familial ALS and
sporadic ALS cases
Mutated TDP-43 can form cellular aggregates
Little is known about normal function, but
seems to play a role in proper RNA metabolism
Since it connects both familial and sporadic
ALS, TARDBP may become a more important
topic of research than SOD1References
Aebischer P, Kato AC. Playing Defense Against Lou Gehrig’s Disease. Scientific American.
2007 Nov: 86-93.
Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation
prevent axonal degeneration. Science 2004 305:1010-1013.
Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein
JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to
astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron.
1997 Feb;18(2):327-38.
Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron
death in ALS. Nat Rev Neurosci. 2001 Nov;2(11):806-19.
Julien JP. ALS: astrocytes move in as deadly neighbors. Nat Neurosci 2007;10:535–537.
Kanekura K, Suzuki H, Aiso S, Matsuoka M. ER Stress and Unfolded Protein Response in
Amyotrophic Lateral Sclerosis. Mol Neurobiol. 2009;39:81–89
Kaspar BK, Frost LM, Christian L, Umapathi P, Gage FH. Synergy of insulin-like growth factor-1
and exercise in amyotrophic lateral sclerosis. Ann Neurol. 2005 May;57(5):649-55.
Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes
expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat
Neurosci. 2007 May;10(5):615-22. Epub 2007 Apr 15.References
Rothstein JD. Current Hypotheses for the Underlying Biology of Amyotrophic Lateral Sclerosis.
Ann Neurol 2009;65 (suppl):S3–S9
Spreux-Varoquaux O, Bensimon G, Lacomblez L, et al. Glutamate levels in cerebrospinal fluid
in amyotrophic lateral sclerosis: a reappraisal using a new HPLC method with coulometric
detection in a large cohort of patients. J Neurol Sci 2002;193:73–78.
Valdmanis PN, Rouleau GA. Genetics of familial amyotrophic lateral sclerosis. Neurology
2008;70;144-152
Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM,
Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu
IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ, Lee VM, Schellenberg GD, Yu
CE. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic
and histopathological analysis. Lancet Neurol. 2008 May;7(5):409-16. Epub 2008 Apr 7.
Xu L, Yan J, Chen D, Welsh AM, Hazel T, Johe K, Hatfield G, Koliatsos VE. Human neural stem
cell grafts ameliorate motor neuron disease in SOD-1 transgenic rats. Transplantation. 2006.
82(7):865–875.
“About ALS.” ALS Association. Sept. 2008. Web. 20 April 2009.
“Amyotrophic Lateral Sclerosis Fact Sheet.” National Institute of Neurological Disorders and
Stroke. National Institutes of Health. April 2003. Web. 20 April 2009.You can also read

















































