Theoretical Study of Vibronic Spectra of Molecule Systems Generated by Photo- and Electronic Excitations
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Theoretical Study of Vibronic Spectra
of Molecule Systems Generated by
Photo- and Electronic Excitations
Ce Song
Licentiate Thesis in Biotechnology
School of Engineering Sciences in Chemistry, Biotechnology and
Health
Royal Institute of Technology
Stockholm, Sweden 2022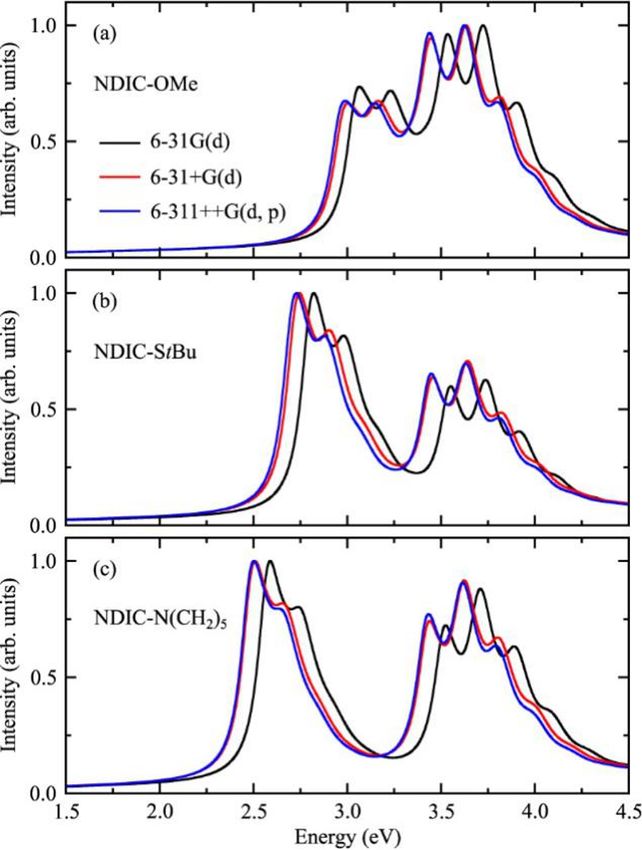
Theoretical Study of Vibronic Spectra of Molecule Systems Generated by Photo- and Electronic Excitations Licentiate Thesis in Biotechnology School of Engineering Sciences in Chemistry, Biotechnology and Health Royal Institute of Technology (KTH) © Ce Song, 2022 ISBN 978-91-8040-149-4 TRITA-CBH-FOU 2022:15 Printed by Universitetsservice US-AB Stockholm, Sweden, 2022
To My Family
Abstract
Spectra represent fingerprints of molecules, which contain unique information
about their properties. Through analyzing the spectral data, one can reveal
the molecules’ energy level alignments, identify their species and geometric
structures, and explore relevant chemical processes and microscopic mechanisms.
Currently, spectroscopy is one of the main means for human beings to enter
the mysterious world of molecules and hear their stories. However, interpreting
molecular spectra is not a straightforward process, because the occurrence of
spectra involves complex interactions between molecules and external stimuli.
Theoretical simulations based on quantum chemistry play an indispensable role in
this regard, which makes developing and applying related computational software
become very important.
This thesis focuses on the theoretical simulations of two types of molecular
spectra, namely the vibrationally resolved optical spectra and the inelastic electron
tunneling spectra (IETS). The former involves the transitions of electrons between
a molecule’s ground state and its excited states with the involvement of molecular
vibrations, and the latter comes from the excitations of a molecule’s vibrational
states within its electronic ground state by inelastic tunneling electrons across a
molecular junction.
By performing time-dependent density functional theory calculations as
well as applying the DynaVib code, I have systematically investigated
the optical absorption properties of two types of functional molecules,
i.e., naphthalenediimide cyclophane (NDIC) derivatives and fused porphyrin
derivatives, which have been proposed as building blocks for future single-
molecule optoelectronic devices. Based on the Franck-Condon (FC) principle,
the simulations well explain the energy shifts induced by chemical substitutions
in the first intense absorption bands of the three NDIC derivatives, and nicely
reproduce the vibrational features of their first two bands. Furthermore, by using
three different exchange-correlation functionals (i.e., the conventional functional
B3LYP and two long-range corrected functionals CAM-B3LYP and ωB97XD),
it is found that long-range corrections are very important for the description of
the spectral features owing to the strong charge transfer in the related excited
states. By taking into account both the FC and the non-FC Herzberg-Teller
(HT) contributions, the experimentally measured electroluminescence spectrum
of a single fused 5,15-(diphenyl)-10,20-(dibromo)porphyrin (fused-H2 P) molecule
is nicely reproduced by the simulations. It is found that the FC contribution also
dominates the emission of the molecule, while the HT terms mainly contribute to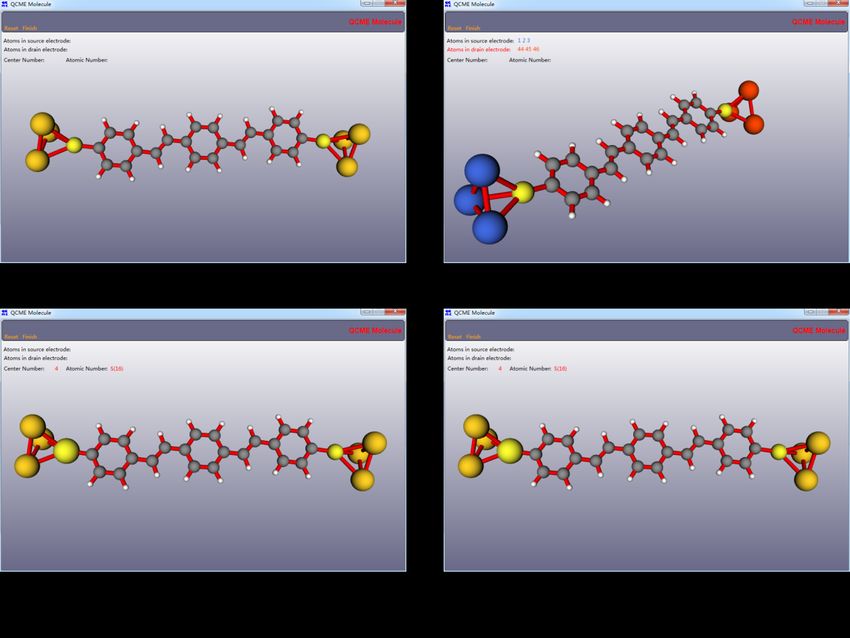
vi
the low-energy tail of the spectrum. The vibrational fine structures as observed
in the experiments are unambiguously assigned based on the simulation results.
In terms of the development of computational software, I have developed a
Windows version for the QCME package − an efficient package to perform first
principles calculations of electron transport through molecules such as simulating
the IETS. The implementation has been achieved by using the C# language and
the Windows Presentation Foundation (WPF) user interface framework. The
Windows version of QCME exhibits compatibility, stability, scalability, and strong
operability. It has a beautiful interface, is easy to learn and to use, and has
improved human-computer interactions. Such an approach of the implementation
can be also extended to other quantum chemistry packages.
Sammanfattning
Spektra representerar molekylära fingeravtryck som innehåller unik
information om deras egenskaper. Genom att analysera spektraldata så
kan man avslöja molekylernas energinivåjusteringar, identifiera deras art
och geometriska strukturer, och utforska relevanta kemiska processer och
mikroskopiska mekanismer. För närvarande är spektroskopi ett av de främsta
sätten för människor att ge sig in i den mystiska världen av molekyler och ta
del av deras berättelser. Det är dock inte en enkel process att tolka molekylära
spektra, eftersom förekomsten av spektra involverar komplexa interaktioner
mellan molekyler och externa stimuli. Teoretiska simulationer baserat på
kvantkemi spelar en oumbärlig roll i detta avseende, vilket gör att utveckling
och tillämpning av relaterad beräkningsprogramvara blir väldigt viktigt.
Denna avhandling fokuserar på de teoretiska simulationerna hos två
typer av molekylära spektra, nämligen den vibrationsupplösta optiska spektra
och den oelastiska elektrontunnelspektra (IETS). Den föregående involverar
elektronövergångar mellan en molekyls grundtillstånd och dess exciterade
tillstånd med involvering av molekylära vibrationer, och den senare uppstår
från excitationerna av en molekyls vibrationstillstånd inom dess elektroniska
grundtillstånd genom oelastisk tunnling av elektroner över en molekylövergång.
Genom att utföra tidsberoende densitetsfunktionella teoriberäkningar
samt tillämpa DynaVib koden så har vi systematiskt undersökt de optiska
absorptionsegenskaperna hos två typer av funktionella molekyler, d.v.s.,
naphthalenediimide cyclophane (NDIC) derivat och fusionerade porphyrin
derivat, varav dessa har föreslagits som byggstenar för framtida enmolekylära
optoelektroniska enheter. Baserat på Franck-Condon (FC) principen så har
simulationerna förklarat energiskiftena inducerade av kemiska substitutioner
i de första intensiva absorptionsbanden hos de tre NDIC-derivaten väl, och
återger vibrationsegenskaperna hos deras två första band på ett snyggt sätt.
Vidare, genom att använda tre olika utbytes korrelationsfunktioner d.v.s., den
konventionella funktionella B3LYP och två långdistans korrigerade funktioner
CAM-B3LYP and ωB97XD), så har man upptäckt att långdistanskorrigeringar
är väldigt viktiga för beskrivningen av de spektrala egenskaperna på grund av
den starka laddningsöverföringen i de relaterade exciterade tillstånden. Genom
att ta hänsyn till både FC och icke-FC Herzberg-Teller (HT) bidrag så kan det
experimentellt uppmätta elektroluminescens spektrumet hos en enstaka fuserad
5,15-(difenyl)-10,20-(dibrom)porfyrin (fused-H2 P) molekylen återges på ett snyggt
sätt av simulationerna. Det visar sig att FC-bidraget också dominerar emissionen
viii
av molekylen, medan HT-termerna huvudsakligen bidrar till lågenergi-änden
av spektrumet. Vibrations finstrukturerna som observerats i experimenten är
tilldelat otvetydig baserat på simuleringsresultaten.
Angående utvecklingen av beräkningsprogramvara så har jag utvecklat
en Windows-version för QCME-paketet - ett effektivt paket att utföra de
första princip beräknelserna av elektrontransport genom molekyler såsom IETS
simulation. Implementeringen har uppnåtts genom att använda språket C#
och användargränssnittet Windows Presentation Foundation (WPF). Windows-
versionen av QCME uppvisar kompatibilitet, stabilitet, skalbarhet, och stark
funktionsduglighet. Den har ett vackert gränssnitt, är lätt att lära sig att använda,
och har förbättrad human-computer interaktioner. Ett sådant tillvägagångssätt
för implementeringen kan även utvidgas till andra kvantkemi-paket.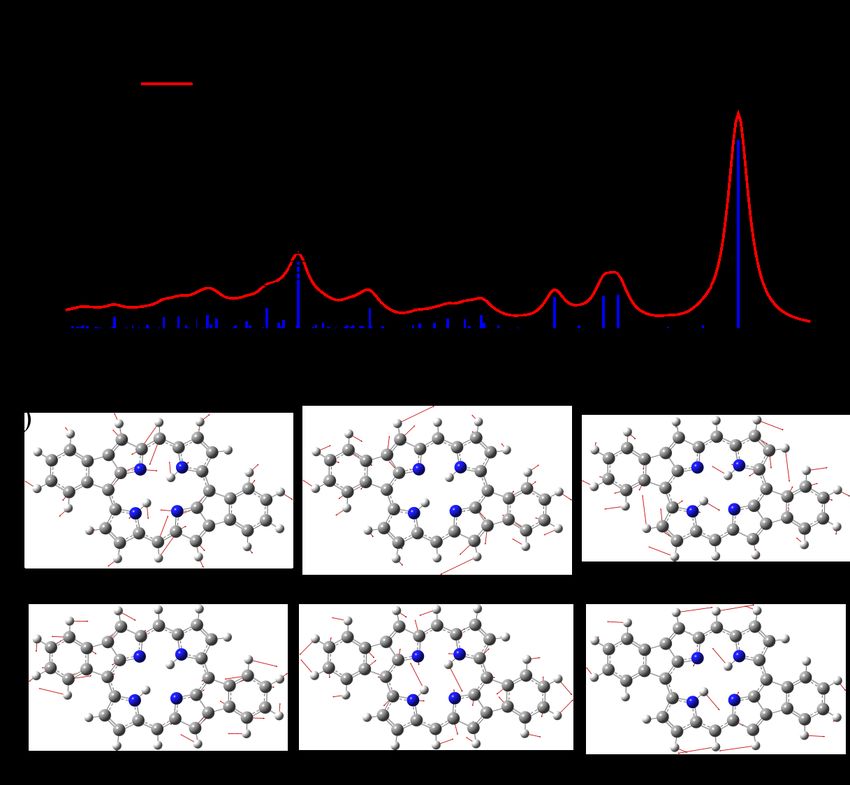
The works presented in this thesis were carried out at the Division of
Theoretical Chemistry and Biology, School of Engineering Sciences in Chemistry,
Biotechnology and Health, Royal Institute of Technology (KTH), Sweden and at
the Hefei National Laboratory for Physical Sciences at the Microscale, University
of Science and Technology of China, Hefei, China.
List of papers included in the thesis
Paper 1. Theoretical simulations for vibrationally-resolved absorption spectra of
naphthalenediimide cyclophane derivatives,
Ce Song, Li Li, Sai Duan, Yi Luo, Guangjun Tian,
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2017,
183, 339-347.
Paper 2. First-principles study on vibrationally resolved fluorescence of fused 5,15-
(diphenyl)-10,20-(dibromo)porphyrin molecule,
Feifei Qiu, Ce Song, Li Li, Yong Wei, Guangjun Tian,
The Journal of Chemical Physics 2018, 149, 074312.
Comments on my contribution to the papers included
All the papers are the results of a teamwork. I am responsible for the
computations and a large part of the writing in paper I. I have contributed to
the discussions and computations in paper II.
List of papers not included in this thesis
Paper 1. Theoretical studies of the structure and properties of anticancer drug taxol,
Meiling Zhang, Ce Song, Zhi Yao, Qiang Ji,
Current Organic Chemistry 2012, 16, 2321-2331.
Paper 2. Comprehensive theoretical studies on the reaction of 1-bromo-3,3,3-
trifluoropropene with OH free radicals,
Meiling Zhang, Ce Song, Yan Tian,
Molecules 2013, 18, 7873-7885.
x
Paper 3. A multiphysics fully coupled modeling tool for the design and operation analysis
of planar solid oxide fuel cell stacks,
Ang Li, Ce Song, Zijing Lin,
Applied Energy 2017, 190, 1234-1244.
Paper 4. Structural information-based method for the efficient and reliable prediction of
oligopeptide conformations,
Xiao Ru, Ce Song, and Zijing Lin,
The Journal of Physical Chemistry B 2017, 121, 2525-2533.
Paper 5. Conformers, properties, and docking mechanism of the anticancer drug
docetaxel: DFT and molecular dynamics studies,
Chuancai Sun, Lijuan Zhu, Chao Zhang, Ce Song, Cuihong Wang, Meiling
Zhang, Yaoming Xie, Henry F. Schaefer III,
Journal of Computational Chemistry 2018, 39, 889-900.
Paper 6. Binding modes of cabazitaxel with the different human β-tubulin isotypes: DFT
and MD studies,
Lijuan Zhu, Chao Zhang, Xudong L, Ce Song, Cuihong Wang, Meiling
Zhang, Yaoming Xie, Henry F. Schaefer III,
Journal of Molecular Modeling 2020, 26, 162.Acknowledgments
It is a great honor to express my acknowledgment to all people who helped
me during my study in Sweden.
First, I would like to express my deepest gratitude to my supervisor, Prof.
Yi Luo, for his professional guidance, continued encouragement, great help, and
strong support. Prof. Luo provides me with an excellent atmosphere for research.
His rigorous spirit in science and keen grasp of research directions have benefited
me a lot. The discussion with Prof. Luo has always been an inspired and enjoyable
experience. I firmly believe that the influence of Prof. Luo on my academic growth
will be long-lasting and continuous.
I also want to thank my co-supervisor, Prof. Yaoquan Tu. I am very grateful
for his valuable advice and warm-hearted help. Even though a long time has
passed, I still remember what Prof. Tu taught me that scientific research cannot
be achieved overnight and it takes time to get to know the little things. The
conversation with Prof. Tu always makes me feel rewarded.
I am very grateful to Prof. Guangjun Tian, who has helped me a lot in
performing theoretical simulations. Guangjun has not only taught me many
specific skills in spectral simulations but also provided me with valuable advice on
how to carry out a scientific project. I am also grateful to guangjun for carefully
reading my thesis and giving very good suggestions for improvement.
Many thanks to Profs. Hans Ågren, Faris Gelmukhanov, Olav Vahtras,
Mårten Ahlquist, and Zilvinas Rinkevicius for their kind discussion and help.
Many thanks to my friends and colleagues Sai Duan, Peng Cui, Li Gao, Xin Li,
Hao Ren, Qiang Fu, Lijun Liang, Wei Hu, Xinrui Cao, Ying Wang, Liqin Xue,
Jiachen Li, Zhengzhong Kang, Bogdan Frecus, Yan Wang, Li Li, Ignat Harczuk,
Weijie Hua, Yuejie Ai, Xiao Cheng, Hongbao Li, Xiaofei Li, Ke-Yan Lian, Vinicius
Vaz da Cruz, Guangping Zhang, Zhen Xie, Yong Ma, Xiuneng Song, Yongfei Ji,
Li-Li Lin, Xing Chen, Junfeng Li, and Lu Sun for their help and all the time we
shared.
Last but not least, I would like to give my sincere gratitude to my wife Yongjin
and my son Chenyu, for their unselfish love and delighted time we shared in the
past, now, and in future.Contents
1 Introduction 1
1.1 Background . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.2 Vibrational spectroscopy . . . . . . . . . . . . . . . . . . . . . . . 2
1.3 DynaVib and QCME . . . . . . . . . . . . . . . . . . . . . . . . . 3
1.4 Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2 Simulation of Vibrationally Resolved Optical Spectra 7
2.1 Theoretical background . . . . . . . . . . . . . . . . . . . . . . . . 7
2.2 Spectral simulations of NDIC derivatives . . . . . . . . . . . . . . 13
2.3 Spectral simulations of fused-H2 P molecule . . . . . . . . . . . . . 18
2.4 Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
3 Simulation of Inelastic Electron Tunneling Spectroscopy 25
3.1 Theoretical background . . . . . . . . . . . . . . . . . . . . . . . . 26
3.2 Object-oriented QCME software . . . . . . . . . . . . . . . . . . . 29
3.2.1 Development environment . . . . . . . . . . . . . . . . . . 29
3.2.2 Description of the QCME calculation on Linux . . . . . . . 30
3.2.3 Design and implementation of QCME on Windows . . . . 30
3.2.4 Extension of the graphic interface to other packages . . . . 35
3.3 Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
4 Conclusions and Future Outlook 39
4.1 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
4.2 Future outlook . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
References 41
xiiiChapter 1
Introduction
1.1 Background
The inclusive and sustainable development of industrialization (ISID) plays a
vital role in improving the living standards of all people worldwide and in
overcoming the environmental and energy challenges faced by modern society. [1]
Large-scale, integrated, and continuous industrial production is the key driving
force for solving the poverty problem and promoting common prosperity. An
indispensable guarantee for the industrialization processes is the accurate and
convenient examination of products. For this purpose, detection technologies
based on spectroscopy have been employed more and more widely.
Structural identification is one of the most vital issue in chemistry, since once
the information of a chemical structure is obtained, all corresponding physical,
chemical and biological properties of the compound can be determined. The
identification of structures thus provides a unique, long-lasting and clear way
of expressing chemical compounds. [2] The spectral analysis developed by German
chemist Bunsen and physicist Kirchhoff in 1859 [3] is one of the effective approaches
for structural identification. As each structure of a substance has its own specific
spectral characteristics, the relationships established between the structures and
the spectra constitute the basis for identifying structural information according
to the spectra.
According to the mechanism of generation, a spectrum can be classified
into emission spectrum, absorption spectrum, and scattering spectrum. Based
on the corresponding wavelength range, it can be divided into X-ray spectrum,
ultraviolet-visible spectrum, infrared spectrum, and Raman spectrum, etc. Among
them, the characteristics of vibrational spectra have significant correlations
12 1 Introduction
with the configurations of molecules. Besides, even in the electronic spectra
of molecules, vibrational features can also be captured (Figure 1.1). Thus,
vibrational spectra play an important role in detecting the structures and
properties of molecules.
Figure 1.1 Schematic diagram of vibration features in electronic spectrs
1.2 Vibrational spectroscopy
Molecular vibration, one of the intrinsic properties of molecules, corresponds to
the periodic back and forth movements of constituent atoms. The generated
vibrational spectrum originates from transitions between different vibrational
levels in the same electronic state. Vibrational spectrum constitutes an important
branch of spectral analysis. Infrared spectrum, Raman spectrum, and sum-
frequency generation, to name a few, belong to this category.
Infrared spectroscopy is widely used in qualitative studies of mixed systems.
This method, however, suffers from the shortcoming of cluttered signals, since
signals from the background cannot be avoided. External environment and the
solvents may contaminate the signal of the target molecule, making it difficult
to distinguish and strip the desired information. Raman spectroscopy can
complement with the infrared spectrum in identifying specific structural features
or characteristic groups, but the weak signal intensity and the low sensitivity
had limited its application for a long time. Fortunately, Raman spectroscopy
has regained the favor of researchers thanks to the advancement of the optical
technology, and two new experimental methods are thus developed, i.e., the
surface-enhanced Raman spectroscopy and the resonant Raman spectroscopy. The
former utilizes surface plasmon and the corresponding signals are greatly affected1.3 DynaVib and QCME 3
by the substrate, while lasers are required in the latter for selectively exciting
vibrations to increase the intensities of the spectra. As another means of the
vibrational spectrum analysis, sum frequency spectroscopy is often used to reveal
the microscopic information of the interface, being a powerful and versatile tool in
surface science. This approach has its shortcomings too, like the different spectral
features appearing under different polarization combinations, for example.
When studying the adsorption and emission spectra that involve transitions
between different electronic states, vibrational fine structures are the key
information in interpreting the high-resolution spectra. Theoretical simulations
of the vibrational fine structural features play an essential role in understanding
the geometric and electronic properties of the target molecules. From the
calculated vibrational fine structures, one can not only compare them with
the vibrational-resolved absorption/emission spectra from the experiments for
substance identification but can also reveal detailed mechanisms about the
molecules at the microscales. It thus provides a practical way to assist the
experimental analysis and provide further information that cannot be easily
obtained merely from experiments. The vibrational-resolved absorption/emission
spectroscopy is considered as one of the most commonly used methods in spectra
analysis.
With the rapid development of molecular electronics, much effort has been
devoted to the fabrication, measurement and artificial control of molecular
devices. [4–9] Here, precisely tuning the contacting and bonding patterns between
functional molecules and electrodes is greatly important, since a very small change
in the molecular configuration may significantly affect the performance of the
constructed device. However, identifying the structural feature is challenging at
the atomic scales. In this context, the inelastic electron tunneling spectrum (IETS)
is extremely useful in identifying configurations of molecular devices, especially
via systematic comparison between experimental measurements and theoretical
simulations.
1.3 DynaVib and QCME
DynaVib [10] and QCME [11] are two packages for the simulations of vibration-
related spectra. Based on the harmonic approximation, the DynaVib code can be
used to simulate vibrationally-resolved optical spectra of polyatomic molecules.
QCME is an efficient tool for simulating elastic and inelastic charge transport4 1 Introduction
properties of molecular junctions. These two packages have been employed in
studying a wide range of molecular systems and have been demonstrated to work
very well.
However, there is still room for improvement when using these two packages.
To be specific, for DynaVib, it is not easy to achieve convergence of the simulated
spectra when the time-independent method is used. To address this issue, we
have developed a revised version of DynaVib in which a more efficient time-
dependent method is implemented. We have also considered the contribution
of the Herzberg-Teller (HT) part in the new version to further enhance the
accuracy of the simulations. As a demonstration, we have simulated the
vibrationally-resolved absorption spectra of the naphthalenediimide cyclophane
(NDIC) molecules and the electroluminescence spectra of fused 5,15-(diphenyl)-
10,20-(dibromo)porphyrin (fused-H2 P) molecule by using the newly developed
package.
Developing a graphical user interface (GUI) for a package will be great helpful
for users to conveniently use the corresponding tool. Here, we have put our
attempt and effort in the QCME package. Concretely, we have developed an
easy-to-use interface for QCME under the Windows system and transplanted the
QCME code to the same operating system. The Windows version of QCME
possesses the advantages of compatibility, stability, scalability, beautiful interface,
easy to learn, easy to use, and strong operability for improved human-computer
interactions.
1.4 Summary
The aim of this thesis is to illustrate the importance of the two types of
vibrational spectra, the vibrationally-resolved optical spectra and the IET
spectra, in identifying molecular configurations. Through the development and
transplantation of the corresponding codes and the investigation of the underlying
mechanisms involving specific molecular systems, the applications of the two
types of spectra are discussed. By developing a graphical user interface for
the simulation code, it is convenient to implement and transplant some typical
quantum chemistry software from Linux to the Windows operating system. The
contents of the following chapters are organized as follows.
In Chapter 2, theoretical backgrounds and applications of DynaVib are
provided. In this chapter, the time-dependent method and the Herzberg-Teller1.4 Summary 5
part will be discussed. The implementation of the code will also be introduced,
followed by an application to the NDIC derivatives and the fused-H2 P molecule.
In Chapter 3, I will give the theoretical backgrounds of IETS and introduce
how the QCME code is developed and transplanted to the Windows operating
system using the C# language and the Windows Presentation Foundation (WPF)
user interface framework. I will also present how to use the Windows version of
QCME.
Chapter 4 includes conclusion and future outlook. We think that the resonant
Raman scattering could be a new feature of the DynaVib software. A portable
graphical user interface of DynaVib, like a mobile app for example, is considered
and could be further developed.Chapter 2
Simulation of Vibrationally Resolved
Optical Spectra
Vibrationally-resolved absorption and emission spectra of molecules can be
simulated by using the DynaVib code. [10] Especially, both the time-independent
sum-over-state method and the time-dependent eigenstate free method have been
implemented, making DynaVib an efficient tool to simulate the spectroscopic
properties of molecules. In this chapter, I will first introduce the theoretical
background, and then present the applications of the DynaVib code, in
investigating the optical absorption properties of a series of naphthalenediimide
cyclophane (NDIC) derivatives and the optical electroluminescence properties of
the fused 5,15-(diphenyl)-10,20-(dibromo)porphyrin (fused-H2 P) molecule. The
NDIC molecules and the fused-H2 P molecule are suitable research objects in
scanning tunneling microscope based single-molecule optical characterizations and
have been demonstrated to exhibit interesting optical responses. [12,13] Detailed
theoretical analysis on the corresponding optical properties of such systems could
be beneficial for the design of single-molecule optoelectronic devices. The large
sizes and the interesting optical properties of the two types of molecules also enable
us to check the accuracy and the practicability of the DynaVib code.
2.1 Theoretical background
In the simulations of the vibrationally-resolved absorption and emission spectra
with DynaVib, the key is to obtain the vibrational profiles, which can be calculated
either by directly computing the vibrational integrals or by evaluating the time-
evolution of the appropriate time-correlation functions. In this section, I will
78 2 Simulation of Vibrationally Resolved Optical Spectra
illustrate the processes by using one-photon absorption as an example. Other
types of optical processes, like the one-photon emission and the multi-photon
nonlinear processes, can be treated in a similar way.
The incident frequency-dependent absorption intensities of an one-photon
absorption process can be written as Eq. 2.1 [14–17]
4π 2 ω X γ
σ (ω) = P (i, T ) |⟨ψi |µ̂| ψf ⟩|2 , (2.1)
3c i,f (ω − ωif )2 + γ 2
where ω represents the frequency of the incident light and c is the speed of light.
|ψi ⟩ and |ψf ⟩ are the wave functions of the initial and the final states, respectively.
ωif is the energy difference between the above two states. P (i, T ) represents the
temperature (T ) dependent Boltzmann population of the initial state |ψi ⟩. Here,
the line shape broadening is described by a Lorentzian function with γ being
the half-width at half-maximum (HWHM). In practice, the broadening can also
be exerted by using other types of functions such as the Gaussian or the Voigt
schemes, depending on the actual requirements. The essential part of Eq. 2.1
is to calculate the transition dipole moment ⟨ψi |µ̂| ψf ⟩ with µ̂ being the dipole
operator. For practical calculations, the Born-Oppenheimer (BO) approximation
and the harmonic approximation need to be employed to obtain the vibrational
transition dipole moment. According to the BO approximation, the electronic
and the nuclear parts of the wave functions can be separated and thus, we have
|ψi ⟩ = |ψie ⟩ |ψiv ⟩ and |ψf ⟩ = ψfe ψfv . Here, the superscripts e and v represent the
electronic and the nuclear part, respectively. Accordingly, the transition dipole
moment can be re-written as Eq. 2.2
⟨ψi |µ̂| ψf ⟩ = ψiv ψie |µ̂| ψfe ψfv , (2.2)
where µeif = ψie |µ̂| ψfe is the electronic part of the transition dipole moment
which is dependent on the nuclear coordinates. However, the nuclear coordinates
dependence of µeif makes it inefficient to directly calculate the integrals on the
right side of Eq. 2.2. To address this issue, it is helpful to expand µeif into a
Taylor series at the equilibrium position (Q0 ) of the initial state |ψi ⟩:
N
X ∂µeif
µeif = µeif (Q0 ) + Qk + · · · · · · , (2.3)
k=1
∂Qk
where N is the number of the vibration modes. It is worth noting that the well-
known Condon approximation is equivalent to merely keeping the first term on2.1 Theoretical background 9
the right side of Eq. 2.3. In this case, the electronic transition dipole moment is
assumed to be independent of the nuclear coordinates.
By substituting Eq. 2.2 into Eq. 2.3, we can get the following expression
N
X ∂µeif
⟨ψi |µ̂| ψf ⟩ = µeif (Q0 ) ψiv | ψfv + ψiv |Qk | ψfv + · · · · · · . (2.4)
k=1
∂Qk
Here, the first term on the right side is the Franck-Condon (FC) term, while
the second one is the Herzberg-Teller (HT) term. In most cases, these two terms
are adequate in the calculations and thus we will neglect higher order terms in
the following discussions.
After substituting Eq. 2.4 into Eq. 2.1, we can obtain the expression for the
one-photon absorption process considering both the FC and the HT terms as
N
2
X ∂µeif
|⟨ψi |µ̂| ψf ⟩|2 = µeif (Q0 )2 ψiv | ψfv + µeif (Q0 ) ψiv | ψfv ψiv |Qk | ψfv
k=1
∂Qk
N
N X
X ∂µeif ∂µeif
+ ψiv |Qk | ψfv ψiv |Ql | ψfv . (2.5)
k=1 l=1
∂Qk ∂Ql
From the right hand side of Eq. 2.5, it can be found that there are three
parts in the absorption spectrum: The first one only contains the FC integral; the
second term includes both the FC and the HT integrals (often referred to as the
FC/HT part); and the third term only contains the HT integrals (often referred
to as the HT part).
Imitating the form of Eq. 2.5, Eq. 2.1 can be rewritten as
δ (ω) = δ F C (ω) + δ F C/HT (ω) + δ HT (ω) , (2.6)
where
4π 2 ω X 2 γ
δ F C (ω) = P (i, T ) µeif (Q0 )2 ψiv | ψfv , (2.7)
3c i,f (ω − ωif )2 + γ 2
N
F C/HT 4π 2 ω X X
e
∂µeif v v γ
δ (ω) = P (i, T ) µif (Q0 ) ψ |ψ ψiv |Qk | ψfv ,
3c i,f k=1
∂Qk i f (ω − ωif )2 + γ 2
(2.8)
and10 2 Simulation of Vibrationally Resolved Optical Spectra
N X N
4π 2 ω X X ∂µeif ∂µeif v γ
δ HT (ω) = P (i, T ) ψi |Qk | ψfv ψiv |Ql | ψfv .
3c i,f k=1 l=1
∂Q k ∂Q l (ω − ωif )2 + γ 2
(2.9)
Eq. 2.6 is the final form for calculating the line intensities within the
framework of the harmonic approximation. As aforementioned, there are two
approaches, the time-independent and the time-dependent ones, that can be used
for the calculation. Considering that detailed discussions have been given in many
previous works [18] regarding the time-independent method, here we mainly focus
on the time-dependent approach.
In contrast to the time-independent method, in which one needs to calculate
the vibrational integrals and perform sum-over-state among a large number of
involved vibrational transitions, in the time-dependent method, one converts the
sum-over-state in Eq. 2.6 into the Fourier integrals of the corresponding dipole
correlation functions. In this way, heavy computations of the vibrational integrals
in the former method can be avoided. Upon the Fourier transformation, Eq. 2.6
can be written as [16]
Z ∞
FC 4π 2 ω e 2
δ (ω) = µ (Q0 ) dtexp[i (ω − ωif ) t]δ F C (t) exp(−γt), (2.10)
3c if 0
N
∂µeif
Z ∞
F C/HT 4π 2 ω X e
δ (ω) = µ (Q0 ) Re dtexp[i (ω − ωif ) t − γt]δ F C/HT (t),
3c k=1 if ∂Qk 0
(2.11)
and
N N
4π 2 ω X X ∂µeif ∂µeif
Z ∞
HT
δ (ω) = Re dtexp[i (ω − ωif ) t − γt]δ HT (t). (2.12)
3c k=1 l=1 ∂Qk ∂Ql 0
Here, δ F C , δ F C/HT , and δ HT are the Franck-Conon, the Franck-
Condon/Herzberg-Teller, and the Herzberg-Teller parts of the absorption cross
section, respectively. An analytical form for the δ F C (t) term has been provided
by Yan and Mukamel [19] as
δ F C (t) = |ψ (t)|−1/2 exp DT f (t) D ,
(2.13)
where
1
ψ (t) = (C+ S ′ A− + C− SA+ ) (C+ SA− + C− S ′ A+ ) , (2.14)
42.1 Theoretical background 11
and
−1
f (t) = −S ′ A− (C+ S ′ A− + C− SA+ ) C− , (2.15)
with
A± = (n̄ + 1) ± n̄exp(iω ′ t),
C± = 1 ± exp(iωt),
−1
ℏω ′
n̄ = exp( )−1 ,
kT
−1/2
ωi
J −1
Sij = ,
ωj′ ij
Di = (ωi )1/2 −J −1 K i ,
and
T
S ′ = S −1 .
Here, J and K are the Duschinsky rotation matrix and the displacement
vector, respectively. ω ′ and ω are the vibrational frequencies of the initial state
and the final state, respectively.
Shuai et al. formulated the analytical expressions for δ F C/HT and δ HT . [16] In
F C/HT
their method, an auxiliary column matrix Hk is defined to obtain δ F C/HT (t),
as in the formula below:
F C/HT
Hk = [01 . . . 1k . . . 02N ]T1×2N . (2.16)
What follows, δ F C/HT (t) can be expressed as
n o
F C/HT T −1
δ F C/HT (t) = −δ F C (t) (Hk ) L F . (2.17)
For the HT term, a square matrix GHT
kl is introduced:
011 012 · · · 01N +1 ···
021 022 · · · 02N +1 · · ·
GHT
kl · · ·
= ··· ··· ··· · · ·
. (2.18)
0k1 0k2 · · · 1kN +1 · · ·
··· ··· ··· ··· ···12 2 Simulation of Vibrationally Resolved Optical Spectra
Thus, the HT term can be rewritten as
n T HT −1 o
−1 −1
δ HT (t) = δ F C (t) iℏTr GHT
kl L + L F Gkl L F , (2.19)
where
" #
B −A
L= , (2.20)
−A B
2N ×2N
and
T
F = K T EJ K T EJ
1×2N
, (2.21)
with
A = af + J T ai J,
B = bf + J T bi J,
and
E = b i − ai .
Here, ai,f is the diagonal matrix for the initial electronic states:
ωi,f k
ai,f k (τ ) = , (2.22)
sin(ℏωi,f k τi,f )
and bi,f is that for the final electronic states:
ωi,f k
bi,f k (τ ) = . (2.23)
tan(ℏωi,f k τi,f )
The above approach has been implemented in DynaVib, [10] which enables
us to simulate vibrationally-resolved absorption and emission spectra very
efficiently. In the following, I will apply DynaVib to investigate the experimentally
observed changes of the optical absorption properties of NDIC derivatives
induced by chemical substitutions and the vibrational fine structure in the
electroluminescence spectra of the fused-H2 P molecule.2.2 Spectral simulations of NDIC derivatives 13
2.2 Spectral simulations of NDIC derivatives
Cyclophanes are hydrocarbons composed of one or more interconnected aromatic
units. Due to their special chemical structures and wide applications, cyclophanes
have attracted the attention of many researchers. [20] NDIC molecule belongs to
a type of special cyclophane compounds consisting of two NDI molecules in a
face-to-face pattern. NDIC derivatives have also been synthesized via group
substitutions for various research purposes. [21–24] For example, Gabutti et al.
synthesized three new NDIC derivatives to perform scanning tunneling microscope
(STM) induced fluorescence measurements. [25,26] In the STM experiments, the use
of such double-layer NDIC derivatives eliminates the need of spacers since one of
the two NDI chromophores can be used to separate the other from the STM
substrates. This self-decoupling property made the NDIC type of molecules quite
appealing for STM luminescence studies which has attracted the attention of many
researchers. [25,27,28]
The core-substituted NDIC derivatives exhibit rich photo-absorption and
emission properties that are dependent on the type of the substituent groups.
Gabutti et al. experimentally studied three NDIC derivatives of NDIC-
OMe, NDIC-StBu, NDIC-N(CH2 )5 , which were produced via substitutions by
dimethoxy, tert-butylsulfanyl and dipiperidinyl, respectively. [26] It was found that
upon the substitution, the first (low-energy) absorption band exhibits a significant
redshift, while there is almost no change in the position of the second band. In
this way, they had achieved chemically tunable Förster resonance energy transfer
(FRET) within the three NDIC derivatives. [26]
We have theoretically investigated the geometric and optical properties of
the three derivatives. Our simulations reproduced the vibrationally-resolved
absorption spectra of the three molecules. The accuracy of the methods and
the DynaVib implementation have been well verified. This theoretical work
may help to shed light on the behaviors of the NDIC derivatives in the STM
induced luminescence experiments as well as in the corresponding energy transfer
processes.
First principles calculations were performed to simulate the vibrationally-
resolved absorption spectra of the three NDIC derivatives, NDIC-OMe, NDIC-
StBu and NDIC-N(CH2 )5 , for a comparison with the experiments. [26] According
to the crystal structure of the NDIC molecule, [25] the two planes of the NDI
components are not parallel but exhibit a certain inclination. For NDIC-StBu, [26]
by contrast, the two planes are almost parallel. Thus, we constructed a tilted14 2 Simulation of Vibrationally Resolved Optical Spectra
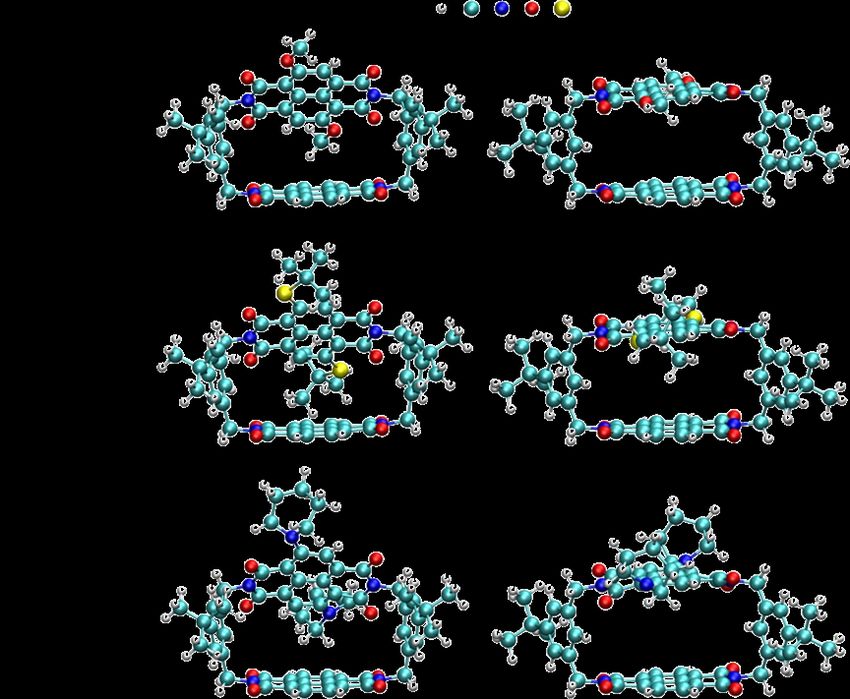
Figure 2.1 Optimized non-planar (left) and planar (right) structures of NDIC-OMe
©
(a), NDIC-StBu (b), and NDIC-N(CH2)5 (c). Reused with permission from ref. 34.
Copyright 2017 Elsevier.
structure (called non-planar structure) and an almost parallel structure (called
planar structure) by replacing the corresponding H atoms and the -StBu groups,
respectively. In Figure 2.1, we present the structures of all the three NDIC
derivatives. The geometries of these molecules at their respective ground states
were optimized within the framework of density functional theory (DFT) by
employing three exchange correlation functionals B3LYP, [29–31] CAM-B3LYP, [32]
and ωB97XD. [33] The 6-31G(d) basis set was used to expand the wave functions.
We have confirmed that the optimized structures correspond to local minima
on the potential energy surfaces since no imaginary frequency was found in the
vibrational analysis.
Since it is computationally very expensive to perform vibrational analysis on
the excited states of large molecules, the linear coupling model (LCM) [35,36] was
employed to simulate the vibrationally-resolved spectra of the NDIC derivatives.
By using the LCM model, one can efficiently obtain the vibrationally-resolved
absorption and emission spectra of large-sized molecules. [36–39] LCM neglects the
Duschinsky mode mixing effect and assumes the potential energy surfaces in the
excited states have the same curvature as those in the ground state and they differ2.2 Spectral simulations of NDIC derivatives 15
with each other only by a shift of the equilibrium positions (the displacement
vector K). Within the harmonic approximation, one can obtain the displacement
vectors from the excited state potential energy gradient at the stable geometries
of the ground states and thus, avoid the computationally demanding vibrational
analysis on the excited states. In practical calculations, only the forces, oscillator
strengths and the excitation energies of the excited state are required which
we obtain with the time-dependent density functional theory (TDDFT). The
polarizable continuum model (PCM) was used to implicitly consider the solvent
effects. The first-principles calculations were carried out by using the Gaussian
09 software package, [40] while the spectral simulations were performed with the
DynaVib code. [10]
The optimized structures of the three NDIC derivatives both in vacuum and
in the dichloromethane solvent were obtained by using different DFT functionals,
with the corresponding results shown in Table 2.1. One can see that for all the
three derivatives, the non-planar structures are more stable than the planar ones.
Both the long-range correction and the empirical dispersion have a big effect on
the calculated results.
Table 2.1 Relative energies (eV) of the three NDIC molecules obtained using different
©
functionals. ∆E is the energy difference (eV). Reproduced with permission from ref.
34. Copyright 2017 Elsevier.
We then calculated the excitation energies and the corresponding oscillator
strengths for the first three excited states of the non-planar structures of the
NDIC derivatives by using the TD-DFT method, with the results shown in Table
2.2. One can see that the excitation energies obtained with the B3LYP functional16 2 Simulation of Vibrationally Resolved Optical Spectra
Table 2.2 Vertical excitation energies and oscillator strengths (in the parentheses)
©
of the three NDIC molecules (non-planar) obtained using different functionals.
Reproduced with permission from ref. 34. Copyright 2017 Elsevier.
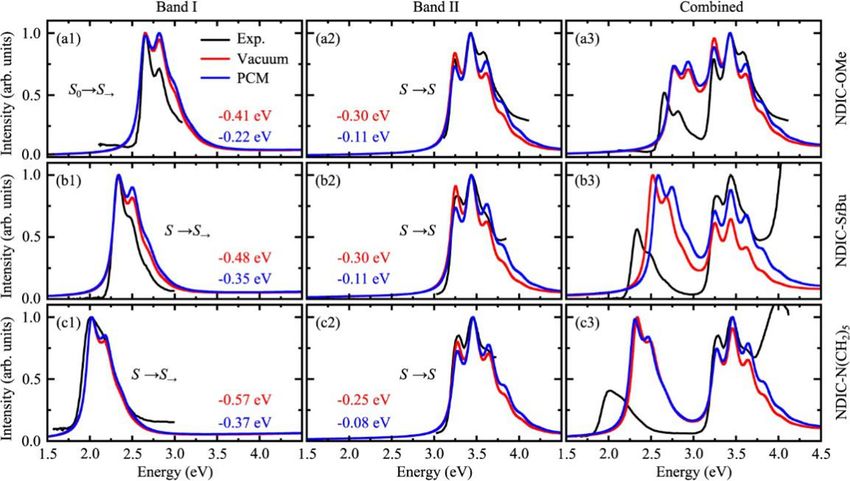
Figure 2.2 Vibrationally-resolved absorption spectra of the three NDIC derivatives.
From top to bottm: NDIC-OMe, NDIC-StBu, NDIC-N(CH2 )5 . The absorption spectra
in the low energy band (band I), the high energy band (band II) and the total absorption
spectra were shown from left to right. The theoretical spectra were shifted to match
the experimental ones with values indicated in the figure. For the combined spectra,
©
the energy shift is the same as those in band II. Reused with permission from ref. 34.
Copyright 2017 Elsevier.2.2 Spectral simulations of NDIC derivatives 17
are significantly lower than the corresponding results obtained from the other
two functionals. Moreover, the first excited states predicted by B3LYP are
only weakly allowed with oscillator strengths, much smaller than those obtained
with the other functionals where the long-range correction were included. In
the experiment, it has been shown that all the three molecules have strong
absorption and emission at the low energy regime, [26] which strongly indicates
that the first excited states are allowed transitions. Therefore, we applied the
CAM-B3LYP and ωB97XD functionals in the simulations of the vibrationally-
resolved absorption spectra. According to the calculation results, it was found
that for all the three molecules, NDIC-OMe, NDIC-StBu, and NDIC-N(CH2 )5 ,
their first excited states exhibit significant redshifts while the positions of the
second absorption band are almost unchanged. By substituting different groups,
a chemically tunable FRET was achieved. [34] Our theoretical simulations nicely
reproduced the observed phenomenon in the experiment. [25,26]
The vibrationally-resolved absorption spectra of the three NDIC derivatives
were simulated by using the CAM-B3LYP and the ωB97XD functionals. Since
the first two absorption bands are both strongly allowed transitions, only the
FC contributions were considered in the simulations. In Figure 2.2, we present
the results for the non-planar structures using the CAM-B3LYP functional. One
can see that the vibrational features of the absorption spectra (the left and the
middle panels), especially those in the first absorption band, are very similar to
the experimental results. There are some differences in the peaks of the first and
the second absorption bands, which may be caused by the core substitution effect.
From the right panel of Figure 2.2, it can be found that differences still
appear between the total simulated absorption spectra and the corresponding
experimentally measured ones. Here, the red shift of the first absorption band is
underestimated with the intensity overestimated, and the intensity of the second
absorption band is underestimated. We have investigated the influence of the
basis set on the simulated spectra, and the corresponding results are shown in
Figure 2.3. One can see that changing the size of the basis sets can slightly affect
the excitation energy. The changes to the energy shift between the two absorption
bands, however, are not obvious. Moreover, adding diffuse functions in the basis
set basically does not cause obvious changes in the simulated spectra. This
indicates that changing basis sets could not improve the comparison between the
simulations and the experiments. Advanced quantum chemistry methodologies
and more accurate solvent models could be needed to further improve the quality
of the simulated vibrationally-resolved absorption spectra.18 2 Simulation of Vibrationally Resolved Optical Spectra
©
Figure 2.3 Simulated absorption spectra of the three NDIC derivatives obtained using
different basis sets. Reused with permission from ref. 34. Copyright 2017 Elsevier.
In short, using the DynaVib code, we have simulated the vibrationally-
resolved spectral profiles at the Franck-Condon level for the first two intense
absorption bands of three NDIC derivatives. Good agreements between the
simulated spectra and the experimental ones are obtained, from which we have
reproduced the vibrational fines structures and explained the electronic origins of
the substitution induced energy shifts observed in the experiments. These results
demonstrate the practicality of the time-dependent method implemented in the
DynaVib code in simulating the vibrationally-resolved optical properties of large
sized molecules.
2.3 Spectral simulations of fused-H2P molecule
Porphyrin and its derivatives have a wide range of applications due to their
unique geometrical and optical properties. Especially, porphyrin molecules have
become the most commonly studied model systems in STM based single-molecule
optical measurements. By combining the advantages of the ultra-high spatial
resolution from STM and the chemical information recognition from optical2.3 Spectral simulations of fused-H2 P molecule 19
characterizations, such type of measurements have significantly enriched our
understanding at the single-molecule level in many fundamental physical and
chemical processes. Meanwhile, the experimental advances also call for more
theoretical efforts in detailed analysis of measured single-molecule spectra, which
plays a key role in uncovering the experimental phenomena.
One interesting example is to analyze the STM induced single-molecule
electroluminesence spectra of the fused-H2 P molecule as measured by Chong et
al. [13] The obtained emission spectra have ultra-narrow peaks with a full-width-
at-half-maximum down to 2.5 meV and exhibiting rich vibronic features. These
results make fused-H2 P an excellent system for spectral simulations because, on
one hand, the experiments were performed using a single-molecule decoupled
from the environment, which is ideal for comparison with theoretical simulations
that employ free molecular models. On the other hand, the rich vibrational
features also serve as a valuable reference for testing different vibrational models.
Especially, unlike the NDIC molecules where the spectra were dominated by the
Franck-Condon parts, the Herzberg-Teller contributions are expected to play an
important role in the emission spectra of porphyrin derivatives and therefore,
should be taken into account in the spectral simulations. In this section, I
will present our work on the theoretical simulations of the vibrationally-resolved
emission spectra of the fused-H2 P molecule.
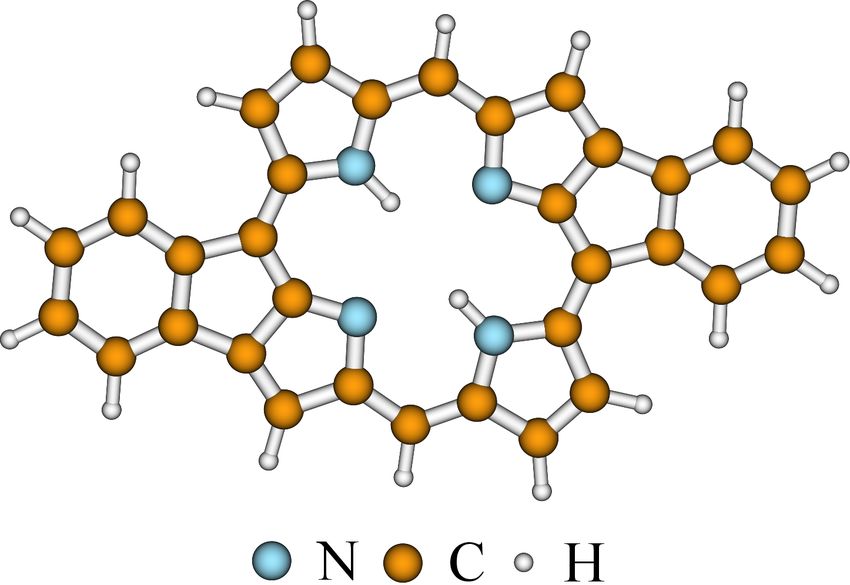
Similar to the study of NDIC molecules, we also performed DFT/TDDFT
calculations to obtain the simulated spectra of fused-H2 P. Thanks to the relatively
smaller size of the fused-H2 P molecule (shown in Figure 2.4), both the ground and
the first excited states were optimized. Frequency analysis were also performed for
the both two states to confirm that stable geometries had been obtained. Three
density functionals, B3LYP, [29–31] ωB97X-D, [33] and M06-2X [41] were used in the
calculations. Our test calculations showed that increasing the size of the basis set
(6-31G(d,p) in the current simulations) did not bring a significant improvement
in the simulated emission spectra.
In the spectral simulations, both the FC and the HT parts were included.
The Duschinsky mode mixing effect was also considered. The derivatives of
the transition electronic dipole moment with respect to the vibrational normal
modes were obtained from the analytical nuclear derivatives that can be calculated
using the Gaussian 16 [40] software after the vibrational calculations at the excited
state [42] . Such the analytical derivatives eliminated possible errors that may
appear in numerical differential approach and thus, further improved the accuracy
of the HT part of the spectra. All the spectra were simulated using the time-20 2 Simulation of Vibrationally Resolved Optical Spectra
©
Figure 2.4 Geometrical structure of the fused-H2 P molecule. Reused with permission
from ref. 42. Copyright 2018 AIP Publishing.
independent method as implemented in the DynaVib software. Such a method
was efficient in simulating the fused-H2 P molecule, which allowed a convenient
assignment to the fine structures of the spectra.
In Figure 2.5, we compare the fluorescence spectra of the fused-H2 P molecule
simulated using the three density functionals with the electroluminescence spectra
of the molecule reported by Chong et al. [13] . It can be immediately noticed that
the three functionals give quite similar spectral profiles, indicating the simple
valence transition feature of the S1 → S0 transition. By comparing the simulated
and the measured spectra, it can be found that the most obvious discrepancy
is the relative intensity of the vibrational peaks. For example, the intensities
of the peaks located at about 1.33 eV and 1.46 eV are overestimated in the
simulated spectra. However, all the experimentally observed vibrational structures
in the electroluminescence spectrum have been nicely reproduced in the simulated
spectra, as indicated by the vertical dashed lines. The good agreement between
the simulated and the experimental spectra indicates that the applied method is
sufficient for the description of the emission process of the fused-H2 P molecule.
An interesting aspect of the porphyrin derivatives is that the HT parts could
also have significant contributions to the optical properties of the molecules. For
example, the optical absorption and emission spectra of the simplest porphyrin
derivatives, porphine (H2 P), were dominated by the HT terms. [43,44] To examine
the HT effect on the emission spectra of the fused-H2 P molecule, we have taken
the simulated spectra using ωB97X-D as the example and compared the total
spectrum with those obtained by considering only the FC or the HT contributions.
The results are demonstrated in Figure 2.6. It can be found that, similar to
the case of the NDIC molecule, the fluorescence of the fused-H2 P molecule is2.3 Spectral simulations of fused-H2 P molecule 21
Figure 2.5 Vibrationally-resolved fluorescence spectra of the fused-H2 P molecule as
simulated with B3LYP (red line), ω B97X-D (green line) and M06-2X (blue line). The
experimental spectrum (black line) from ref. 13 was also shown for comparison. The
©
simulate spectra were blue shifted by 0.27 eV, 0.10 eV, and 0.09 eV for the three
functionals, respectively. Reused with permission from ref. 42. Copyright 2018 AIP
Publishing.
also dominated by the FC part. The HT part only contributes to the low-
energy tail of the spectrum. Such an interesting change can be attributed to
the geometrical changes from free H2 P type to the fused configuration in fused-
H2 P. In fact, symmetry analysis shows that the S1 → S0 transition is a dipole
allowed Bu → Ag transition with major contributions coming from the lowest
unoccupied molecular orbital (LUMO) to the highest occupied molecular orbital
(HOMO), as shown in the inset of Figure 2.6. This also confirms the valence
nature of the emission transition and further explains the good performance of all
the three density functionals.
The good agreement between the simulated and the measured spectra enables
us to make a detailed assignment to the vibrational fine structures in the emission
spectra of the fused-H2 P molecule. Figure 2.7 (a) shows such a detailed plot
where we have included both the simulated (red) and the measured (black) spectra
together with the bar spectrum of the FCHT factors as computed by the time-
independent method using the DynaVib software. It can be found that the most
intense vibrational peaks are the 0-1 vibrational transitions of different modes.
The lack of high order transition in the emission spectra can be attributed to
the relatively small FC activity of the totally symmetric modes of the fused-H2 P
molecule. The broad peaks in the emission spectra, such as the one around 1.4 eV,
are a collective contribution from several nearby transitions. It is interesting to
see that the simulated spectra actually capture such detailed features quite nicely,22 2 Simulation of Vibrationally Resolved Optical Spectra
Figure 2.6 Detailed analysis of the fluorescence spectra of the fused-H2 P molecule as
obtained with the ω B97X-D functional. Black line is the total spectrum containing both
the FC (red) and HT (blue) contributions. The electronic transition between LUMO
©
and HOMO, which dominates the emission process, is shown as the inset. Reused with
permission from ref. 42. Copyright 2018 AIP Publishing.
demonstrating the accuracy of the applied method.
The six most active modes corresponding to the intense vibrational peaks
in the emission spectra can be found in Figure 2.7 (b). It can be seen
that all the six modes are in-plane modes mainly involving atoms in the
porphyrin chromophore. This is also part of the reasons for the good agreement
between the simulated fluorescence spectra and the experimentally measured
electroluminescence spectrum for which the fused-H2 P molecule was decoupled
from the metallic electrodes through the terthiophene side chains. This was
further verified in our calculations, where it was found that the inclusion of
the terthiophene side chain have negligible influences on the simulated emission
spectra [42] .
In this section, I have presented our simulations on the optical emission
spectra of the fused-H2 P molecule. It is found that the three used density
functionals (B3LYP, ωB97Xd, and M06-2X) give quite similar spectral profiles
while the ωB97Xd and M06-2X functionals improve the obtained excitation
energy by 0.15 eV. Test calculations with different basis set indicates that the
standard 6-31G(d,p) basis set is sufficient to describe the optical properties of
the fused-H2 P molecule. The simulated spectra are in good agreement with
the experimentally measured single-molecule electroluminescence spectra of the
molecule, which enables us to a give detailed assignment to the vibrational origins
of the fine spectral structures. It has also been found that the high energy part of2.4 Summary 23
Figure 2.7 (a) Vibrational assignment of the fluorescence spectra of the fused-H2 P
molecule. Black and red lines are the experimental spectrum and the simulated
spectrum, respectively. The blue bar spectrum is the calculated FCHT factors. (b)
©
Active vibrational modes for the main emission peaks in the fluorescence spectra.
Reused with permission from ref. 42. Copyright 2018 AIP Publishing.
the emission spectra of the molecule is dominated by the 0-1 FC transitions and
the HT part only contributes to the low energy tail of the spectra.
2.4 Summary
This chapter is devoted to the theoretical simulations of vibrationally-resolved
molecular optical spectra. The theoretical background, especially the theory of the
time-dependent method for the simulation of the vibrationally-resolved electronic
spectra is presented. This method, which has been implemented in the DynaVib
software developed by our group, is then applied for two types of functional
molecules, namely NDIC derivatives and fused-H2 P molecule. The simulated
spectra are used to explain some interesting experimental features reported for
the two types of molecules, which is not only helpful for our understanding of the24 2 Simulation of Vibrationally Resolved Optical Spectra experimental results but also facilitates the future application of such molecules in the design of single-molecule optoelectronic devices.
Chapter 3
Simulation of Inelastic Electron
Tunneling Spectroscopy
With the aim of building ultra-small electronic devices, molecular electronics
has attracted tremendous research interests in the past two decades. Great
advancements have been achieved in the experimental realization of functional
molecular devices like switches and rectifiers. [8,45–47] However, since the measured
electronic signals in the current-voltage or the conductance-voltage characteristics
lack chemical information, it is not straightforward to determine whether a
molecule really exists, and if so, how the electron transport properties across
the junction are affected by the behavior of this molecule.
Inelastic electron tunneling spectroscopy (IETS), [7] which corresponds to the
second derivative of the current with respect to the bias voltage, is an ideal tool
to address the above issue. Generally, there are two types of charge transport
processes in molecular junctions: the elastic tunneling and the inelastic tunneling
(Figure 3.1). Since the inelastic tunneling can be triggered via vibrational
excitations of molecules within the junctions due to the coupling between
the tunneling electrons and the molecular vibrational motions, the vibrational
properties of the sandwiched molecules can be reflected by IETS.
Compared with conventional vibrational spectroscopic techniques, such as
infrared absorption or Raman scattering, IETS has different selection rules. [48]
This makes IETS be an promising characterization tool for capturing vibration
modes that are invisible in conventional vibration spectra. [49] Intriguingly, IETS
measurements can be conducted in real space at the single-molecular scale,
empowering IETS the ability to provide the real space distribution of vibrational
modes with sub-angstrom resolutions. The single-molecular scale measurement
25You can also read

















































