Sarah Christine Bening - Exploring and Enhancing Context-Dependent Beta-Lactam Antibiotic Efficacy
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Exploring and Enhancing Context-Dependent Beta-Lactam Antibiotic Efficacy by Sarah Christine Bening B.Bm.E., University of Minnesota (2015) Submitted to the Department of Biological Engineering in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biological Engineering at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY February 2021 © Massachusetts Institute of Technology 2021. All rights reserved. Author . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Department of Biological Engineering December 29, 2020 Certified by . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . James J. Collins Professor of Biological Enginneering Thesis Supervisor Accepted by . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Katharina Ribbeck Professor of Biological Engineering Chair of Graduate Program, Department of Biological Engineering
2
Exploring and Enhancing Context-Dependent Beta-Lactam Antibiotic Efficacy by Sarah Christine Bening Submitted to the Department of Biological Engineering on December 29, 2020, in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biological Engineering Abstract Antibiotics, such as beta-lactams, are essential medical tools for the treatment of bacterial infections. Unfortunately, clinical treatment efficacy is declining over time as bacteria adapt to and evade antibiotic treatment through mechanisms called an- tibiotic resistance, tolerance, and persistence. Antibiotic tolerance and persistence, in particular, are often context-dependent phenotypes: environmental factors can in- fluence bacterial physiology and alter antibiotic efficacy. Optimal antibiotic use, as well as strategies to enhance antibiotic efficacy, can therefore be informed by studies of context-dependent antibiotic action. In this thesis, I present three vignettes about beta-lactam antibiotic efficacy and how environmental context alters in vitro treatment outcomes. First, I explore bac- terial killing in multi-drug contexts, focusing on how different beta-lactams can have different effects in combination with antibiotics of other classes. Second, I present a new counter-tolerance method using metabolic stimulation to sensitize tolerant, stationary phase bacteria to beta-lactam antibiotics. Third, I present an extension of this metabolic counter-tolerance strategy, now combining metabolic and target- specific stimulation to further enhance beta-lactam efficacy. I demonstrate that this combined approach, when coupled with beta-lactamase inhibitors, restores beta- lactam sensitivity to simultaneously tolerant and resistant cultures of clinically rele- vant pathogens. I conclude by discussing opportunities for future study into antibiotic context-dependence and the application of counter-tolerance approaches such as the one described in this thesis. Thesis Supervisor: James J. Collins Title: Professor of Biological Enginneering 3
4
Acknowledgments My parents made sure I loved reading. There were always books in the house for me to read. While I read a lot of books growing up, somewhere around high school or undergrad I stopped finding the time to read for fun. Thankfully, shortly after my thesis proposal, I started reading books again. There have been so many good books that have helped me relax and get through the tough parts of graduate school, so many opportunities to go on someone else’s adventure for a while. Unlike when I was younger, my reading habits during graduate school always started with a flip to the acknowledgments section. I wanted to see what circles the author ran in (and maybe find some new books to read). More importantly, the acknowledgments were a chance to hear the author’s voice, unfiltered by their characters. A chance to hear from the real actual human that created each story. You’d think after reading all those acknowledgment sections writing this would’ve been a little easier. Anyways, here’s my thank you to all the people who helped me on my graduate school adventure: To Jim – when I started graduate school I knew next to nothing about how to study bacteria or about how antibiotics work. Thank you for giving me the opportunity to learn, the support and resources to make mistakes, and the freedom to pursue the projects that interested me. To my committee, Katharina Ribbeck and Mike Laub – thanks for your support and encouragement, and thank you for taking the time and energy to really be present at my committee meetings. I always enjoyed talking science with you. To the Collins lab – with no exceptions that I can think of (though there may very well be some), everything I know about microbiology research at the bench I learned from you all. I’m grateful for this massive and varied group of researchers that I got to work with and learn from every day. To the group from my early days in the Collins lab – Prerna Bhargava, Rebecca Shapiro, Caroline Porter, Saloni Jain, and Meagan Hamblin – thank you for helping me get started. Thank you for helping me learn how to be a scientist, how to work hard, ask questions, and celebrate the good experiments. And thank you for your 5
friendship, for all the lunch chats, snack breaks, trips to the Muddy, and recently the never ending group chats. To the Collins lab graduate students – especially those at the Broad: Ian Andrews, Bernardo Cervantes, Meagan Hamblin (honorary graduate student), and Erica Zheng – you’ve been a great group to struggle through graduate school with. And I mean struggle in the best way: a good struggle’s okay. You’ve been a great group to learn with, work hard with, and fail miserably at making random dinner restaurant decisions with. To the group that are coauthors on the core of my thesis work – Ian Andrews, Meagan Hamblin, and Allison Lopatkin – thank you for being my closest collabora- tors. Thank you for the experiments you did, but more importantly, thank you for all the time we spent talking science and for helping me learn how to present this story. To the MIT BE community – especially BE 2015, the BE Grad Board, the BE Communication Lab, and the BE department staff – I don’t know how to say it better than "community." The different groups I’ve been part of have given me the opportunity to get to know and work with so many more people than the typical graduate student might, and I’ve benefitted so much from getting to know you all. I’m grateful to be part of a community that works hard and knows how to have fun. To my friends – classmates, roommates, coworkers, friends from before MIT and outside of MIT – thank you for all the time we had fun and weren’t stressed about science. For all the juggling, cookie balling, moth fighting, Bopping to the Top, Muddy-ing, Naco Taco-ing, and (unfortunately) Zooming times. Thanks for giving me the space to just be a human and stop thinking about PCR for a while. Thanks for your friendship and helping me get across the finish line. To my sister, Dr. Bening – thank you for always setting the bar high. You’re right: I definitely had it easy never having to take the bus to high school. Thank you for being my best and longest teammate and friend. To my parents – thank you for everything. Growing up you made sure I had the opportunity to do everything I wanted to do. And all those things – all the sports, (recovering from) all the knee injuries, all the piano lessons and everything trombone, 6
and all the people I’ve gotten to know along the way – have made me who I am today. Thank you for your example of loving to read, learn, and work hard. Thank you for the environment you created at home, and for making sure school was never a scary thing. It’s because you encouraged me to keep chasing what I was interested in that I’m writing my PhD thesis acknowledgments. 7
8
Contents 1 Introduction 13 1.1 Overview of Thesis Chapters . . . . . . . . . . . . . . . . . . . . . . . 13 1.2 Antibiotic Failure Modes . . . . . . . . . . . . . . . . . . . . . . . . . 14 1.3 Antibiotic Efficacy and Context-Dependence . . . . . . . . . . . . . . 15 1.4 The Beta-Lactam Antibiotics and Bacterial Peptidoglycan . . . . . . 17 2 Characterization of Killing by Bactericidal Antibiotic Combinations 21 2.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21 2.2 Synergy with Aminoglycosides is Common . . . . . . . . . . . . . . . 23 2.3 Concentration-Dependent Interactions Between Beta-Lactams and Amino- glycosides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25 2.4 Exploring Non-Beta-Lactam Cell Shape Perturbations . . . . . . . . . 27 2.5 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29 2.6 Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 3 A Metabolic Counter-Tolerance Strategy for Beta-Lactams 33 3.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33 3.1.1 Targeting Metabolism as a Counter-Tolerance Strategy . . . . 34 3.1.2 Peptidoglycan Activity in Stationary Phase . . . . . . . . . . 35 3.2 Stationary Phase Bacteria are Highly Tolerant to the Beta-Lactam Ampicillin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35 3.3 Many Carbon Sources Restore Killing by High Ampicillin Concentrations 37 3.4 Cell Growth May Contribute to but Does Not Explain Sensitization . 40 9
3.5 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45 4 Enhancing Beta-Lactam Counter-Tolerance Using D-Amino Acids 49 4.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49 4.1.1 Stationary Phase Peptidoglycan . . . . . . . . . . . . . . . . . 50 4.1.2 L,D-Transpeptidases and Beta-Lactam Efficacy . . . . . . . . 50 4.1.3 D-Amino Acids and Beta-Lactam Efficacy . . . . . . . . . . . 51 4.2 Testing Single D-Amino Acids . . . . . . . . . . . . . . . . . . . . . . 52 4.3 Combining D-Amino Acids with Metabolic Stimulation by Carbon Sources . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55 4.4 Application to Other Organisms and 100% LB . . . . . . . . . . . . . 60 4.5 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64 5 Future Directions and Conclusions 69 5.1 Antibiotic Discovery and Resistance . . . . . . . . . . . . . . . . . . . 69 5.2 Context-Dependence and Clinical Relevance . . . . . . . . . . . . . . 70 5.3 Application of Counter-Tolerance Approaches . . . . . . . . . . . . . 72 5.3.1 Context-Dependent Carbon Source Efficacy . . . . . . . . . . 72 5.3.2 Delaying the Evolution of Antibiotic Resistance . . . . . . . . 73 5.3.3 Failure Modes of Counter-Tolerance Strategies . . . . . . . . . 74 A Methods for Sensitizing Tolerant Bacteria to Beta-Lactam Antibi- otics 91 10
List of Figures 1-1 Antibiotic Efficacy is a Systems-Level Process that is Sensitive to External Cues. . . . . . . . . . . . . . . . . . . . . . . . . . . . 16 2-1 Killing by Low Concentrations of Quinolones or Beta-Lactams in Combination with Sublethal Gentamicin. . . . . . . . . . . . 23 2-2 Synergy of Select Beta-Lactams at Low Concentrations with Additional Aminoglycosides. . . . . . . . . . . . . . . . . . . . . . 24 2-3 Effects of Aztreonam and Mecillinam Treatment on E. coli Cell Shape. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25 2-4 Aztreonam and Gentamicin Synergy. . . . . . . . . . . . . . . . 26 2-5 Mecillinam and Gentamicin Antagonism. . . . . . . . . . . . . . 26 2-6 Concentration-Dependent Interactions of Ciprofloxacin and Penicillin with Gentamicin. . . . . . . . . . . . . . . . . . . . . . 27 2-7 Gentamicin Combinations with A22 or SulA Overexpression. 28 3-1 Stationary Phase E. coli are Highly Tolerant to the Beta- Lactam Ampicillin. . . . . . . . . . . . . . . . . . . . . . . . . . . 36 3-2 Many Carbon Sources Sensitize Stationary Phase E. coli to Ampicillin. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37 3-3 Ampicillin with Single Carbon Sources Has Limited Efficacy Against Stationary Phase E. coli. . . . . . . . . . . . . . . . . . 38 3-4 Sensitization to Ampicillin by Xylose Depends Upon Xylose Concentration. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39 11
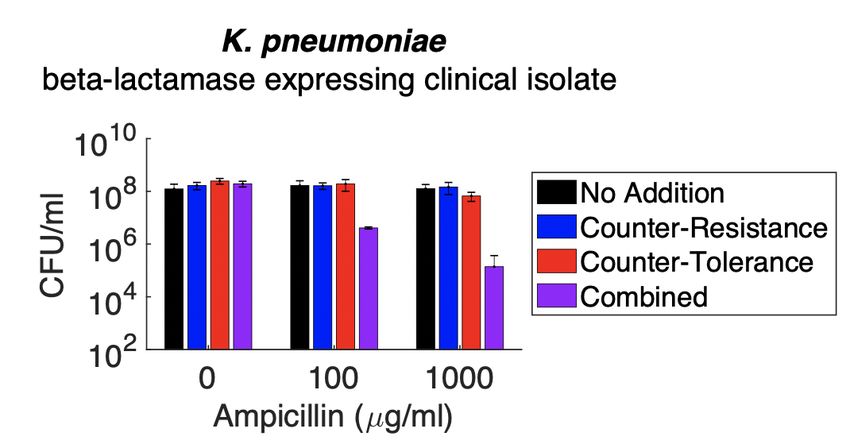
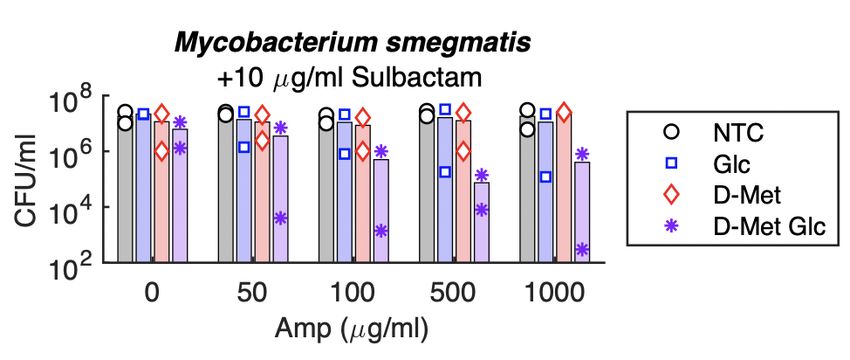
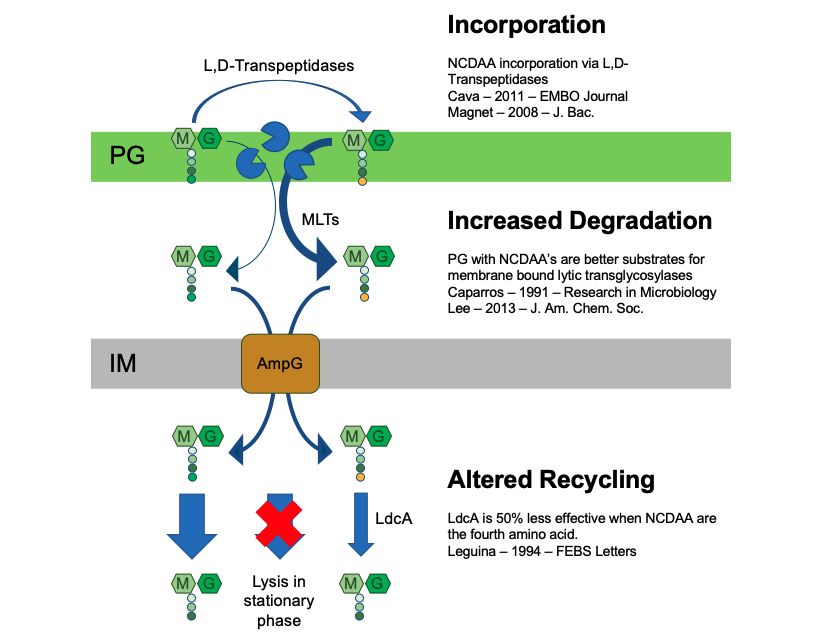
3-5 Carbon-Stimulated Growth in 1% LB is not Correlated with Effectiveness at Stimulating Lysis by Ampicillin . . . . . . . . 41 3-6 Carbon Sources do not Stimulate Population-Level Growth in 100% LB. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42 3-7 Fluorescence Dilution Measurement of Single-Cell Growth of Glucose or Xylose Treated Cultures. . . . . . . . . . . . . . . . 44 4-1 Most D-Amino Acids Alone Are Ineffective at Sensitizing Ampicillin Even in Low Density Cultures. . . . . . . . . . . . . 53 4-2 D-Alanine Affects Ampicillin Sensitivity . . . . . . . . . . . . . 54 4-3 Combining Pyruvate with D-Alanine is Not Optimally Effec- tive in WT E. coli . . . . . . . . . . . . . . . . . . . . . . . . . . . 56 4-4 D-Amino Acids Potentiate Ampicillin when Combined with Pyruvate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57 4-5 D-Methionine Specifically Potentiates Beta-Lactam Antibiotics. 59 4-6 D-Methionine Still Enhances Ampicillin Lethality in a ∆metNIQ Knockout. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60 4-7 Xylose and D-Methionine Enable Killing by sub-100 g/ml Beta-Lactam Concentrations. . . . . . . . . . . . . . . . . . . . . 61 4-8 Fluorescence Dilution Measurement of Single-Cell Growth of D-Methionine Treated Cultures. . . . . . . . . . . . . . . . . . . 62 4-9 Sensitizing Stationary Phase K. pneumoniae to Ampicillin . 63 4-10 Sensitizing Stationary Phase M. smegmatis to Ampicillin . . 65 4-11 Effects of Non-Canonical D-Amino Acids (NCDAAs) on As- pects of Peptidoglycan Maintenance . . . . . . . . . . . . . . . . 67 12
Chapter 1 Introduction 1.1 Overview of Thesis Chapters In this dissertation, I describe three vignettes about beta-lactam antibiotic efficacy, exploring both how beta-lactams work in different contexts and how we can manip- ulate environmental contexts to make beta-lactams more effective. In Chapter 1, I provide motivation for understanding and improving antibiotic efficacy as it relates to preserving the clinical usefulness of antibiotic therapy. I describe the recent work which reveals the complex, context-dependent aspects of antibiotic action, and provide relevant background on the beta-lactam antibiotics and their target, bacterial peptidoglycan. Beta-lactam antibiotics are not only a major class of antibacterials for clinical use, but have also played a significant historical role in experimental studies of antibiotic action. In Chapter 2, I describe work exploring the lethal outcomes of combinations of bactericidal antibiotics, featuring antibiotics from the beta-lactam, quinolone, and aminoglycoside classes. In particular, I focus on the beta-lactam antibiotics and explore how different beta-lactams can have different outcomes in combination with other drugs. In Chapter 3, I describe a metabolic strategy to enhance beta-lactam lethality against stationary phase E. coli through the addition of a single carbon source. I characterize the concentration-dependence of this strategy and describe how previous 13
studies did not observe beta-lactam lethality in similar conditions. In Chapter 4, I build upon the strategy described in Chapter 3 and describe a target-specific strategy to further enhance beta-lactam lethality against stationary phase bacteria. I provide evidence that D-amino acids enhance beta-lactam efficacy through interacting with bacterial peptidoglycan, and apply this combined strategy – carbon source and D-amino acid supplementation – to sensitize different bacteria in stationary phase to beta-lactam antibiotics. In Chapter 5, I discuss the current state of antibiotics research and future direc- tions specific to understanding and utilizing context-dependence to improve antibiotic efficacy. 1.2 Antibiotic Failure Modes Antibiotics are of great clinical importance for the treatment of bacterial infections. Unfortunately, antibiotic treatment can fail for multiple reasons, even when an antibi- otic is able to reach an infection site. Specifically, bacteria can phenotypically evade antibiotic action using three major mechanisms: antibiotic resistance, tolerance, and persistence [16]. Antibiotic resistance is a phenotype – often driven by genetic changes – which enables bacteria to grow in high concentrations of an antibiotic. Antibiotic tolerance and persistence, in contrast, describe reduced killing efficacy of an antibi- otic against a bacterial culture, resulting in a reduced population-level (tolerance) or sub-population (persistence) killing rate by the antibiotic. The clinical relevance of antibiotic resistance is well recognized [22]. We have es- tablished methods for quantifying the resistance level of infectious organisms, we know much about common mechanisms of resistance [33], and counter-resistance strategies such as antibiotic cycling or resistance-targeting adjuvants are being developed and deployed clinically [130, 37, 62, 107, 40]. In contrast, less is known about the mechanisms and clinical relevance of both antibiotic tolerance and persistence, though there is increasing evidence of these phe- nomena clinically in the form of chronic and recurrent infections [86, 47]. Treatment 14
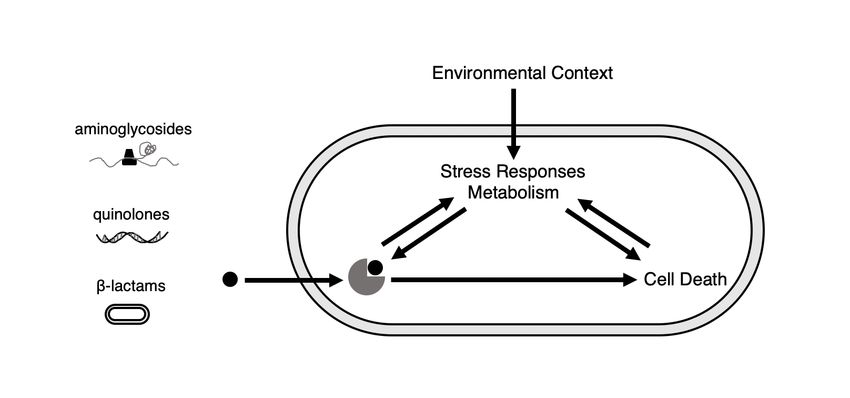
methods aimed at increasing antibiotic lethality against tolerant or persistent bacteria are beginning to be developed [86]. Importantly, recent work has demonstrated how the reduced killing efficacy of antibiotics due to tolerance or persistence can facilitate the evolution of antibiotic resistance [128, 73, 75]. Collectively, these antibiotic failure modes threaten the current and future abil- ity of antibiotics to treat bacterial infections while also pointing towards important avenues for research meant to preserve antibiotic efficacy. Studying mechanisms of antibiotic action – as well as mechanisms of resistance, tolerance, and persistence – can lead to interventions that improve and preserve antibiotic efficacy both for current antibiotics and those still being developed [18, 112]. In particular, antibiotic efficacy is context-dependent [131], and thus understanding of the environmental factors af- fecting treatment outcomes will inform both clinical stewardship programs and the development of new antibiotics or adjuvants targeting bacterial metabolism, stress responses, or other aspects of physiology in order to enhance antibiotic efficacy [65]. 1.3 Antibiotic Efficacy and Context-Dependence Antibiotic lethality requires downstream processes in addition to target inhibition. The primary targets of most bactericidal antibiotics are involved in essential cellular processes. Quinolone antibiotics inhibit the essential protein DNA gyrase, amino- glycosides bind to the ribosome and inhibit translation, and beta-lactam antibiotics inhibit penicillin binding proteins (PBPs) that are required for peptidoglycan syn- thesis. However, antibiotics do more than inhibit essential cellular processes (Figure 1-1). Quinolone antibiotics form toxic complexes containing broken DNA [82], and beta-lactam antibiotics lead to futile cycling of cell wall synthesis [25, 26]. Several studies have linked off-target cellular damage to antibiotic-mediated cell death, in- cluding the involvement of toxic reactive oxygen species (ROS) and DNA damage [39, 125, 6]. Broad metabolic [6, 135, 77, 36] and transcriptomic [27, 39, 90, 17] per- turbations caused by antibiotics indicate that primary target inhibition is critically linked to bacterial physiology. 15
Figure 1-1: Antibiotic Efficacy is a Systems-Level Process that is Sensitive to External Cues. Antibiotics bind to and inhibit their primary targets (left) which contributes to cell death. Primary target inhibition also influences and is influenced by bacterial metabolic and transcriptomic states, which further impact cell death process and are also susceptible to influence from the extracellular environment. Given these systems-level disruptions, it is apparent then that bacterial physiol- ogy can also impact antibiotic lethality, and thus external factors acting on the cell can alter antibiotic efficacy. In particular, external metabolic conditions can drasti- cally alter antibiotic efficacy. Blocking protein translation or cellular respiration can inhibit the lethality of bactericidal drugs with different targets in both Escherichia coli and Staphlyococcus aureus [77]. TCA cycle activity enhances lethality of the aminoglycoside tobramycin in Psuedomonas aeruginosa, while flux through the gly- oxylate shunt protects bacteria [87]. Additionally, external conditions can act through bacterial metabolism and alter cellular ATP levels. Multiple studies have shown, by varying culture conditions, that the lethality of many bactericidal drugs is dependent upon ATP, with increased lethality occuring at higher ATP levels [79, 136]. En- vironmental factors altering transcriptional states and stress response activity also impact antibiotic efficacy. For example, when antibiotics are used in combinations, transcriptional responses in the cell differ from what is observed with single drug treat- ment, impacting the efficacy of the combination treatment [14]. Continued study into 16
both downstream consequences of antibiotic target inhibition and context-dependent antibiotic efficacy will suggest new methods to improve antibiotic efficacy through altering bacterial physiology. Context-dependent antibiotic efficacy is particularly important for antibiotic tol- erance and persistence. Resistance is predominantly a genetic phenotype [33], and genetic resistance can be assayed in clinical settings through growth-inhibition as- says or sequence-targeted assays [8]. In contrast, environmental conditions such as starvation drastically influence antibiotic lethality [50, 117] and can lead to antibi- otic tolerance and persistence. Unlike the stability of genetic changes, phenotypic or context-dependent antibiotic efficacy is challenging to assay in the clinic [86, 128]. Therefore, continued study of antibiotic efficacy and context-dependence is necessary to improve our ability to identify and combat antibiotic treatment failure due to tolerance and persistence. 1.4 The Beta-Lactam Antibiotics and Bacterial Pep- tidoglycan Since the discovery of penicillin in 1929, the beta-lactam class of antibiotics has been expanded through both natural product discovery and chemical modifications to form a large group of antibiotics with varying spectra of activity [76]. The clinical significance of beta-lactam antibiotics is clear, as in 2009 over half of clinical spending on antibiotics in the United States was spent on beta-lactam antibiotics [54]. New beta-lactam derivatives are continuing to be developed and continue to form part of the preclinical pipeline of new antibiotics [112]. Thus, beta-lactams form an antibiotic class with great current and future clinical relevance, motivating continued study into their mechanisms of action and opportunities for improvement. Beta-lactam antibiotics inhibit synthesis of bacterial peptidoglycan [123, 103, 76]. Bacterial peptidoglycan is a common and effective antibiotic target [103], as peptido- glycan is a normally essential and bacterial-specific polymer [123]. There are many 17
enzymes active in building, maintaining, and degrading bacterial peptidoglycan, some of which will be discussed here and later in chapter 4 (see section 4.1). Importantly, following cytosolic synthesis of peptidoglycan precursor molecules, two key reactions insert new peptidoglycan: glycan polymerization by glycosyltransferases and peptide cross-linking by transpeptidases [123]. Beta-lactams inhibit this latter reaction, as the core beta-lactam structure mimics the terminal D-ala-D-ala dipeptide present in newly synthesized and uncross-linked peptidoglycan [114], and thus beta-lactams are able to bind to and inhibit the DD-transpeptidases known as penicillin-binding pro- teins (PBPs). As peptidoglycan plays a key role in the maintenance of cell shape [123], beta-lactam treatment causes dramatic changes in cell shape, specific to the cellular role of the PBPs targeted by a given drug [46]. Beta-lactams, in addition to their clinical significance, have been key to many scientific discoveries about bacterial physiology and, importantly, bacterial evasion strategies: resistance, tolerance, and persistence. The evolution of and clinical emer- gence of beta-lactam resistance mirrors that observed with other antibiotics: clinical observations of resistance rapidly following – or even preceeding – introduction of a new antibiotic into the clinic [28]. Indeed, penicillin resistance was observed in 1940 even before widespread use of penicillin to treat infections during World War II and afterwards [76]. Further, observations often cited as the first descriptions of antibiotic persistence occurred during the study of beta-lactam action [59, 10]. Re- cent advances in our understanding of tolerance and persistence featured beta-lactam antibiotics, such as early single-cell studies into persister mechanisms [3], demonstra- tion of the evolution of antibiotic tolerance [44], and the demonstration that evolved tolerance can facilitate the evolution of antibiotic resistance [73]. Additionally, con- tributing to the study of bacterial persistence, beta-lactams are frequently used as a mechanism to isolate bacterial persisters. Methods include using a beta-lactam to lyse non-persistent bacteria and enable the collection of non-lysed persisters [64], and alternatively using a beta-lactam which induces filamentation to separate sensitive filamentous cells from small, non-growing persisters using size-based filtration [127]. Tolerance to beta-lactam antibiotics can also be used to isolate auxotrophic mutants, 18
thus contributing to the study of bacterial metabolism [34], and the PBP-specificities of beta-lactam antibiotics make beta-lactams useful chemical genetics tools for the study of bacterial peptidoglycan. As just one of many examples in E. coli, use of the PBP2-specific beta-lactam mecillinam led to the characterization of interactions be- tween peptidoglycan-cleaving endopeptidases and peptidoglycan-synthesizing PBPs [67]. Collectively, these studies and many others demonstrate both the clinical and sci- entific significance of beta-lactam antibiotics. Continued study into the mechanisms of beta-lactam action and methods to enhance beta-lactam activity are thus expected to broadly impact future work in both the clinic and the lab. 19
20
Chapter 2 Characterization of Killing by Bactericidal Antibiotic Combinations 2.1 Introduction Antibiotics are frequently used in combination clinically when the drugs are syner- gistic, when patients are critically ill and require treatment before completing sus- ceptibility tests, or when prolonged treatment may select for resistance, thereby in- creasing the likelihood the infection is susceptible to at least one drug [71]. Results of recent studies have suggested current practices of using synergistic drugs are actually counterproductive in combating antibiotic resistance. These findings support the ar- gument that synergistic interactions increase selection pressure and drive acquisition of resistance, suggesting that antagonistic combinations may be better at preventing selection of resistance [94, 115, 88, 55]. Both of these potentially confounding needs – increasing treatment efficacy and limiting the spread of resistance – depend upon how drugs interact, motivating further understanding of drug interaction mechanisms. Growth inhibition assays enable relatively rapid screening of antimicrobial interac- tions in vitro [23, 132]. While these methods have the benefit of being high through- put, this leaves differences in reported interactions between studies unexplained, as mechanistic followup must be limited. In one recent study, aminoglycosides in com- bination with either beta-lactam or quinolone antibiotics were antagonistic in E. coli 21
[23], while in another these combinations were synergistic [132]. These differences may be due to differences in growth conditions like media richness or the use of different methods to quantify interactions. Given the clinical acceptance of beta-lactam com- binations with aminoglycosides [71], the inconsistencies in these screens is intriguing. Early studies of the lethality of beta-lactam/aminoglycoside combinations in the 60s, 70s, and 80s measured increased uptake of a radiolabeled aminoglycoside when com- bined with a beta-lactam [97, 91, 89]. However, it is unclear why some beta-lactams are antagonistic with aminoglycosides [92] or why synergy isn’t observed in some organisms [89]. Further mechanistic studies of these beta-lactam/aminoglycoside in- teractions may shed light into these discrepancies and could also inform clinical use of these antibiotic combinations. Recent studies have moved beyond observing drug interactions and have taken a mechanism-oriented approach to study antibiotic combinations [77, 90, 23, 14, 24]. These studies used screens of single gene deletion libraries [23, 24], analysis of tran- scriptional responses [90, 14, 24], and perturbations of metabolic consequences [77] to characterize drug interactions. This focus on mechanism enabled testable predictions extending to other drugs or stressors [77, 90, 23, 24] and organisms [23]. Insights into downstream responses of bactericidal antibiotic treatment may similarly be useful to understand combinations of beta-lactam, aminoglycoside, and quinolone antibiotics. In particular, aminoglycoside uptake and activity can be stimulated by metabolites that enhance respiration and membrane potential [87, 2, 95], and bactericidal antibi- otics of the quinolone and beta-lactam drug classes have been shown to stimulate res- piration [77]. Together, this suggests respiration increases stimulated by quinolone or beta-lactam antibiotics could enhance uptake of aminoglycoside antibiotics. Studying how metabolic and transcriptional perturbations induced by bactericidal antibiotics affect outcomes of combination treatment may clarify interaction mechanisms and identify critical environmental factors that tune these interactions. Here, we aim to characterize the killing efficacy of bactericidal drug combinations. By enumerating CFUs and evaluating antibiotic lethality, this work addresses a regime unavailable to high throughput growth inhibition screens. 22
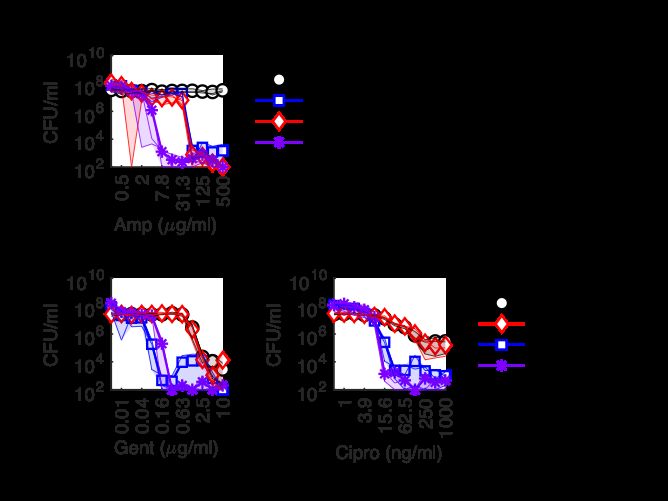
2.2 Synergy with Aminoglycosides is Common To explore combinations of bactericidal antibiotics, we began with antibiotic com- binations with the aminoglycoside gentamicin, motivated by the clinical relevance of beta-lactam/aminoglycoside antibiotic combinations [71]. Uptake of aminoglyco- side antibiotics, such as gentamicin, into the cell is dependent upon proton motive force (PMF) [110]. Based upon previous work that observed increased aerobic res- piration rates in response to bactericidal antibiotics [77], we hypothesized that this antibiotic-induced electron transport chain activity could play a role in combinations of aminoglycosides with other antibiotics. Because norfloxacin-induced increases to bacterial respiration were maximal near the antibiotic’s minimum inhibitory concen- tration (MIC), doses of quinolone and beta-lactam antibiotics were selected to be near the MIC. All quinolones and three of the four beta-lactams tested showed synergy with gentamicin at these concentrations (Figure 2-1). Since gentamicin alone was Figure 2-1: Killing by Low Concentrations of Quinolones or Beta-Lactams in Combination with Sublethal Gentamicin. Percent survival after three hours of treatment. Dotted line marks 100% sur- vival. Quinolones: Ciprofloxacin (Cipro, 20 ng/ml), Levofloxacin (Levo, 30 ng/ml), Nalidixic Acid (Nal, 5 g/ml), Norfloxacin (Nor, 40 ng/ml). Beta-lactams: Penicillin (Pcn, 30 g/ml), Cefsulodin (Cef, 50 g/ml), Aztreonam (Azt, 500 ng/ml), Mecil- linam (Mec, 500 ng/ml). Bars represent the mean with error bars representing the standard error of the mean for 5 replicates, except mecillinam which is 2 replicates. non-lethal, these interactions can be classified as synergy because the lethality of the combination is more than that of the quinolone or beta-lactam alone. In contrast, 23
the combination of the beta-lactam mecillinam with gentamicin led to less killing than observed with mecillinam alone, indicative of antagonism, and consistent with previous studies which showed antagonism between mecillinam and aminoglycoside antibiotics [92]. Given the clinical relevance of beta-lactam/aminoglycoside combinations and fre- quently observed synergy between these drug classes, we also tested combinations of these beta-lactams with other aminoglycoside antibiotics, to see if the observed synergy was consistent in our experimental conditions. Here we did not include mecillinam, which was found to be antagonistic with gentamicin. In this panel of 4 beta-lactams and 3 aminoglycosides, we again found that synergy was common when combining beta-lactams near their MIC and sublethal aminoglycoside concentrations (Figure 2-2). Figure 2-2: Synergy of Select Beta-Lactams at Low Concentrations with Additional Aminoglycosides. Percent survival after three hours of exposure. Beta-lactams: Penicillin (Pcn, 30 g/ml), Cefsulodin (Cef, 50 g/ml), Aztreonam (Azt, 500 ng/ml), Piperacillin (5 g/ml). Aminoglycosides: Gentamicin (Gent, 50 ng/ml), Kanamycin (Kan, 600 ng/ml), Streptomycin (Strep, 500 ng/ml). Bars represent the mean with error bars representing the standard error of the mean for 3 replicates. 24
2.3 Concentration-Dependent Interactions Between Beta-Lactams and Aminoglycosides Additional concentrations were then tested for combinations of the beta-lactams aztreonam and mecillinam with gentamicin. These two antibiotics were chosen be- cause they are known to inhibit complementary processes of peptidoglycan synthesis (Figure 2-3). Mecillinam inhibits PBP2, which is required for cell elongation, and Figure 2-3: Effects of Aztreonam and Mecillinam Treatment on E. coli Cell Shape. thus mecillinam treatment induces sphere formation [104]. Aztreonam, in contrast, in- hibits PBP3, which is required for cell division, and thus aztreonam treatment induces cell filamentation [51]. At the drug concentrations tested, combinations of aztreonam and gentamicin always led to increased killing (Figure 2-4). In contrast, lethal con- centrations of mecillinam where antagonised by sublethal gentamicin (Figure 2-5, top) and lethal concentrations of gentamicin were antagonized by mecillinam (Figure 2-5, bottom). The findings with aztreonam and mecillinam establish a preliminary pattern: syn- ergy between aminoglycosides and filamentation-inducing treatments, and antago- nism between aminoglycosides and sphere-inducing treatments. In order to explore this pattern further, we first wanted to explore concentration-dependent drug in- teractions for other antibiotics with the aminoglycoside gentamicin. To do this, we used the quinolone antibiotic ciprofloxacin as well as the beta-lactam antibiotic penicillin, because both penicillin [48] and ciprofloxacin [84] are known to induce 25
Figure 2-4: Aztreonam and Gentamicin Synergy. Percent survival after three hours of exposure. The data presented in both figures is the same and serves to highlight gentamicin concentration-dependence (left) and aztreonam concentration-dependence (right). Data is presented as the mean of 3 replicates; error bars are standard error of the mean. Figure 2-5: Mecillinam and Gentamicin Antagonism. Percent survival after three hours of exposure. The data presented in both figures is from the same experiment and are separated to highlight combinations with sublethal gentamicin (top) and lethal gentamicin (bottom). Data is presented as the mean of 3 replicates with individual replicates shown. dose-dependent changes in cell morphology. In our experimental conditions, both ciprofloxacin and penicillin were found to have dose-dependent interactions with gen- 26
tamicin (Figure 2-6), where low gentamicin concentrations antagonized killing by higher ciprofloxacin or penicillin concentrations, but lower ciprofloxacin or penicillin concentrations – concentrations near the MIC – were synergistic and showed increased killing with gentamicin. Given the known concentration-dependent morphological Figure 2-6: Concentration-Dependent Interactions of Ciprofloxacin and Penicillin with Gentamicin. Percent survival after three hours of treatment. Left: gentamicin and ciprofloxacin, mean and standard deviation of two replicates. Right: gentamicin and penicillin, a single replicate. changes of both ciprofloxacin and penicillin, these results are supportive of a poten- tial role for morphology in combinations with aminoglycoside antibiotics. However, because concentration-dependent morphological effects were not verified in these ex- perimental conditions, the link between morphology and aminoglycoside interactions is still limited. 2.4 Exploring Non-Beta-Lactam Cell Shape Pertur- bations Because the concentration-dependent morphological effects of ciprofloxacin and peni- cillin complicate analysis without concurrent microscopy studies, we next sought out alternative methods to perturb cell shape which don’t have concentration-dependent effects on cell shape. Two such methods were identified and tested (Figure 2-7A). First, the small molecule A22 inactivates a protein other than PBP2 that is involved 27
in cell elongation, MreB [45]. Similarly to mecillinam, A22 inhibited killing by lethal concentrations of gentamicin (Figure 2-7B). Inducible overexpression of the protein Figure 2-7: Gentamicin Combinations with A22 or SulA Overexpression. A) Schematic depicting expected cell shape changes caused by SulA overexpression and A22 treatment. B) Percent survival after three hours of treatment with gentam- icin and A22 (single replicate, representative of additional experiments not show). C) Percent survival after three hours of treatment with gentamicin and ATC induction of WT or mutant L83R SulA (single replicate, representative of additional experiments not shown). SulA was used as an alternative method to mimic the action of aztreonam and prevent cell division. SulA is induced by the SOS response to inhibit FtsZ polymerization, which is required for cell division [60]. SulA was cloned from the MG1655 genome into a plasmid and placed under the control of an inducible promoter responsive to anhydrotetracycline (ATC). As seen with aztreonam, inhibition of cell division by SulA led to decreased cell viability (Figure 2-7C), an effect not due to protein over- 28
expression, as indicated by the lack of interaction between gentamicin and a mutant form of SulA unable to bind FtsZ [60]. 2.5 Discussion Here I presented work characterizing concentration-dependent lethality of bacterici- dal antibiotic combinations. We find that synergy between both beta-lactam and quinolone antibiotics with sublethal aminoglycoside antibiotics is common, with the exception of the beta-lactam mecillinam. We then characterize dose-dependent inter- actions of various antibiotics with the aminoglycoside gentamicin, and suggest an ini- tial hypothesis that changes in cell shape correlate with gentamicin interactions: that is, filamentation-inducing treatments synergize with gentamicin and sphere-inducing treatments are antagonistic with gentamicin. As a first step towards testing this hy- pothesis, we present data with two other cell-shape perturbations: antagonism with the MreB-inhibiting and sphere-inducing A22, and synergy with overexpression of the FtsZ-inhibitor and thus filamentation-inducing SulA. Further exploration of this shape-related aminoglycoside synergy should initially focus on three next steps. First, microscopy should be incorporated into these stud- ies such that cell shape perturbations – particularly concentration-dependent effects as previously reported with ciprofloxacin [82] and penicillin [48] – can be directly correlated with killing phenotypes in the tested experimental conditions. Second, initial mechanistic studies using radiolabelled or fluorescently labelled aminoglyco- sides should be done, as increased aminoglycoside transport is frequently reported for beta-lactam/aminoglycoside synergy [97, 91, 89]. As with microscopy studies, amino- glycoside uptake studies would allow correlation of killing phenotypes with a potential mechanistic explanation – increased aminoglycoside transport – in identical experi- mental conditions. Finally, the third recommended next step is including additional methods of perturbing cell shape. This can include – but is not limited to – the fol- lowing: single-gene knockouts with altered cell shape including the filamentous holD mutant and spherical rodZ mutant [43]; filamentation from shifts to non-permissive 29
temperatures for temperature sensitive mutants of cell division proteins; cells with altered dimensions due to titrated levels of key elongation (MreB) and division (FtsZ) proteins; plasmid-mediated over-expression of other proteins inhibiting FtsZ (for ex- ample CbtA [57], SlmA [7], or MinC [9]) or MreB (for example CbtA [57] or BolA [42]); and the inclusion of addition beta-lactams antibiotics [46]. While an attractively simple hypothesis that beta-lactam/aminoglycoside inter- actions can be classified according to the effects of the beta-lactam on cell shape, this hypothesis has limitations that should be explored further. First, beta-lactam and aminoglycoside synergy is also noted in spherical bacteria [89]. While beta- lactam treatment still affects cellular morphology in spherical bacteria, the extremes of filamentation and sphere-formation observed in the normally rod-shaped E. coli don’t directly translate to non-rod-shaped bacteria. Thus, extension of a cell shape as a determinant of beta-lactam/aminoglycoside synergy hypothesis to other bac- teria by necessity requires more nuance. Two potential avenues to explore are as follows: to quantify cell surface area or volume rather than shape, and see if these metrics are correlated with aminoglycoside transport; beta-lactam treatment induces other physiologic changes (for example, to bacterial metabolism and stress responses [131]), occuring in parallel to changes in cell shape, and further characterization and perturbation of those physiological changes may be more directly correlated with aminoglycoside synergy than cell shape. A second outstanding question about shape- dependent beta-lactam/aminoglycoside interactions relates to drug interactions in conditions where beta-lactams induce shape changes but are not lethal. For exam- ple, both aztreonam and mecillinam induce shape changes in E. coli when grown in poor growth conditions such as minimal medium. If shape changes determine beta- lactam interactions with aminoglycosides, synergy and antagonism would still occur in these conditions. However, as aztreonam and mecillinam are not lethal in minimal medium, bactericidal processes downstream of primary target inhibition are likely al- tered in these conditions, and if these secondary physiological changes play a key role in beta-lactam/aminoglycoside interactions, drug interactions would change despite consistent changes in cell shape. Thus, in addition to testing additional cell shape 30
perturbations, testing combinations with gentamicin and mecillinam or aztreonam in minimal media or anaerobic conditions should be explored further. Finally, I note that much of this data is preliminary and exploratory, and additional replication and conditions must be completed. 2.6 Methods Reagents: Antibiotics and PBS were purchased from Sigma-Aldrich. MOPS EZ Rich medium was purchased from Teknova. Strains and Plasmids: All experiments were done with E. coli MG1655 or MG1655 Pro (lab stock [39]) in MOPS EZ Rich Medium. SulA was amplified off of MG1655 genomic DNA and cloned into pZE21 using the KpnI and HindIII sites. The SulA L83R mutant was made using overlapping PCR and similarly cloned into KpnI and HindIII. Plasmids were transformed into MG1655 Pro to enable inducible expression; these cultures were grown with 50 g/ml kanamycin for plasmid mainte- nance. Killing Assays: For all experiments, overnight cultures from glycerol stocks were grown in MOPS EZ Rich Medium. In the morning, overnight cultures were diluted 1:5000 in fresh medium and grown to exponential phase, when antibiotics were added and treatment started (T=0, OD600=0.1). Percent survival reflects colony forming units (CFUs) relative to the measured density at T=0. 31
32
Chapter 3 A Metabolic Counter-Tolerance Strategy for Beta-Lactams 3.1 Introduction Both antibiotic resistance and antibiotic tolerance allow bacteria to evade antibiotic action, limiting the effective treatment of bacterial infections [16, 86]. Many mech- anisms of antibiotic resistance are well understood, and strategies such as adjuvants and multidrug combinations targeting these resistance mechanisms are being devel- oped and deployed clinically [130, 37, 62, 107, 40]. In contrast, less is known about antibiotic tolerance — where genetically susceptible bacteria survive typically lethal antibiotic challenge [16]. Tolerant bacteria lead to chronic and costly infections in the clinic [86], and antibiotic tolerance was recently shown to facilitate the evolution of antibiotic resistance in vitro [73] and in the clinic [75]. In order to improve and preserve antibiotic efficacy, treatment strategies combating antibiotic tolerance are beginning to be developed [86]. Here, we aim to develop and characterize a metabolic counter-tolerance strategy for beta-lactams using E. coli. Bacterial metabolism is critically important for antibi- otic efficacy, and metabolism has been successfully targeted to sensitize tolerant E. coli to other classes of antibiotics. Previously, direct translation of these approaches to beta-lactams have not worked with E. coli. Thus, this work aims to fill a gap for 33
the clinically important beta-lactam antibiotics. 3.1.1 Targeting Metabolism as a Counter-Tolerance Strategy Antibiotics and bacterial metabolism interact bidirectionally [105, 131]. Antibiotic treatment leads to broad perturbations in bacterial metabolism, which has been observed in multiple species [6, 36, 135, 134]. Additionally, bacterial metabolic state impacts antibiotic efficacy, such that cellular respiration [77] and ATP levels [136, 79, 102, 29] tune antibiotic lethality. Collectively, this shows that metabolism is an important part of antibiotic efficacy. Bacterial metabolism, which is sensitive to environmental conditions and factors, is thus a logical target for countering antibiotic tolerance. This was first done in 2011, where stationary phase cultures were treated with the quinolone ofloxacin, and survivors – the persister fraction – were then resensitized to aminoglycoside antibiotics through stimulation by individual carbon sources [2]. This approach was found to be efficacious against E. coli and S. aureus [2], E. tarda [95], and P. aeruginosa [87]. This single-carbon strategy also showed efficacy against S. aureus persisters challenged with daptomycin [98]. Unfortunately, the usefulness of this approach was limited, and antibiotics from non-aminoglycoside classes, including quinolones and beta-lactams, were not sensi- tized in those initial studies [2, 95, 87]. Recently, a study found that sensitizing quinolones required a carbon source and an electron acceptor such as oxygen or fu- marate [50]. This was found to work with E. coli, S. aureus, and M. smegmatis [50]. This two part approach with both a carbon source and electron acceptor still did not show efficacy with beta-lactam antibiotics and E. coli. However, this study does highlight that metabolic counter-tolerance strategies may be drug-specific; while the aminoglycoside-developed strategy did not work initially with quinolones, further study with quinolones led to a quinolone-developed strategy that did, suggesting that specific study with beta-lactams may lead to a metabolic counter-tolerance strategy that works with beta-lactam antibiotics. 34
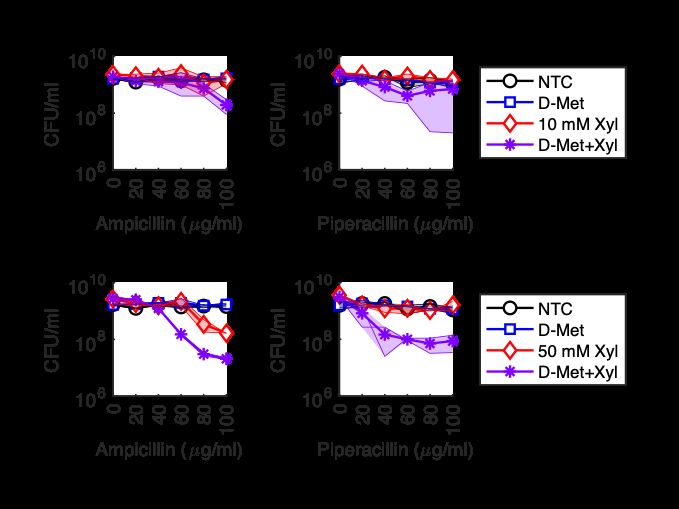
3.1.2 Peptidoglycan Activity in Stationary Phase Beta-lactam antibiotics have drastically reduced effectiveness against stationary phase bacteria, a physiological state commonly associated with phenotypic tolerance to many bactericidal drug classes. Despite high beta-lactam tolerance, peptidoglycan synthesis, maintenance, and turnover processes are still active in stationary phase. New peptidoglycan synthesis is drastically reduced in stationary phase, though it is measurably non-zero [11]. Further, evidence that peptidoglycan turnover and new synthesis are active come from studies of ldcA mutants. The ldcA gene encodes an L,D-carboxypeptidase which processes peptidoglycan turnover products which then are recycled for new insertion into peptidoglycan. The lcdA mutant lyses specifically in stationary phase due to reduced peptidoglycan cross-linking from incorporation of improperly recycled peptidoglycan fragments [111]. Importantly, the peptidoglycan recycling pathway of which LcdA is a part is not essential [93], only LcdA due to aberrant peptidoglycan synthesis in the absence of LcdA. Thus, deletion of lcdA does not lead to lysis because of uncontrolled degradation of peptidoglycan, but rather degradation accompanied by improper synthesis, again pointing towards non-zero peptidoglycan synthetic activity in stationary phase which can serve as a starting point for sensitizing stationary phase E. coli to beta-lactam antibiotics. Given the measurable activity of beta-lactam targets in stationary phase [11], the central role of metabolism in beta-lactam antibiotic lethality, and the presence of drug- and organism-specific differences in other metabolism-directed anti-tolerance strategies (see section 3.1.1), we sought to develop a metabolic strategy to sensitize tolerant stationary phase E. coli to beta-lactam antibiotics. 3.2 Stationary Phase Bacteria are Highly Tolerant to the Beta-Lactam Ampicillin Our experimental protocol consisted of growing bacterial cultures for 24 hours to stationary phase, followed by 24 hours of treatment, after which we measured cul- 35
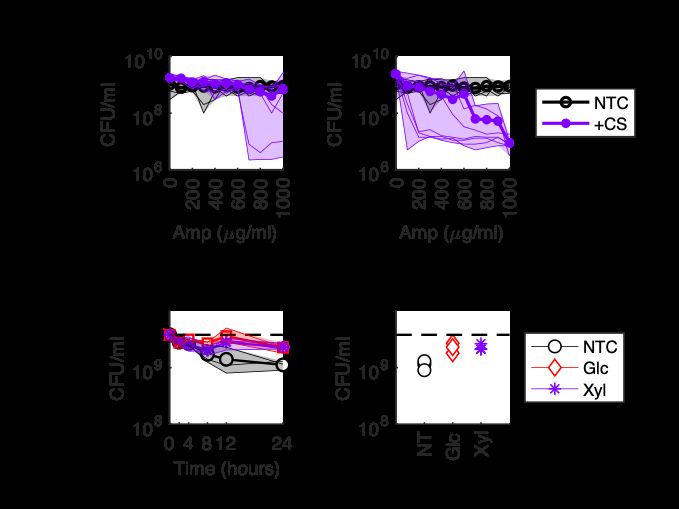
Figure 3-1: Stationary Phase E. coli are Highly Tolerant to the Beta-Lactam Ampicillin. A) Schematic depicting timeline of experiments and corresponding culture density in CFU/ml. B) Survival of stationary phase cultures of MG1655 E. coli grown in 100% (left) and 1% (right) LB and treated with 100 or 1000 g/ml ampicillin (Amp) for 24 hours. Data represents the mean of two replicates with individual replicates shown. ture density (Figure 3-1A). Using this protocol, we confirmed that stationary phase MG1655 Escherichia coli grown in LB medium are tolerant to treatment with 100 g/ml ampicillin (Figure 3-1B), as we have observed previously [50]. We also con- firmed that stationary phase E. coli are tolerant to 1000 g/ml ampicillin (Figure 3-1B), a high tolerance level which has previously been used to study peptidoglycan synthesis and modification in stationary phase [20]. Because beta-lactam efficacy is known to be effected by cell density [41, 63], we next wanted to test if stationary phase tolerance was due to culture density. To do this, we grew stationary phase cultures in LB medium diluted 1:100 in PBS (called 1% LB), reducing the stationary phase cell density by reducing the available nutrients in the culture medium. Cultures grown in 1% LB were also tolerant to 1000 g/ml ampicillin (Figure 3-1B). This suggests that tolerance of E. coli to beta-lactams in stationary phase is not due to culture density but rather growth state. 36
3.3 Many Carbon Sources Restore Killing by High Ampicillin Concentrations Figure 3-2: Many Carbon Sources Sensitize Stationary Phase E. coli to Ampicillin. Survival of stationary phase cultures grown in 100% LB (top) or 1% LB (bottom) treated for 24 hours with 1000 g/ml ampicillin and 10 mM indicated carbon source. Bars are the mean of two replicates with replicates shown. NTC: no carbon con- trol; Glc: glucose; Mann: mannitol; Fruc: frutose; Glyc: glycerol; Gluc: gluconate; Ara: arabinose; Rib: ribose; Xyl: xylose; Pyr: pyruvate; Fum: fumarate; Succ: suc- cinate; Cit: citrate; Ace: acetate; Oxalo: oxaloacetate; Glyoxyl: glyoxylate; Prop: proprionate; Mal: malate. We next tested a panel of carbon sources to see if they could restore killing of stationary phase E. coli by 1000 g/ml ampicillin. Unlike previous studies which tested one metabolic stimulus – optimized for a different antibiotic class – with only 100 g/ml ampicillin [2, 50], we reasoned that using a higher ampicillin concentration, 37
testing a panel of carbon sources, and also testing 1% LB would make it more likely we would find a carbon source that restored beta-lactam lethality. Indeed, we found that in both 1% and 100% LB many carbon sources sensitized stationary phase E. coli to killing by 1000 g/ml ampicillin (Figure 3-2). The carbon sources which showed efficacy in 100% LB belong to glycolysis and the pentose phosphate pathway, whereas carbon sources from the TCA cycle also showed efficacy in 1% LB. Furthermore, a larger log-reduction in culture viability was observed in 1% LB compared to 100% LB. Overall, these results demonstrate that single carbon sources are able to sensitize tolerant stationary phase E. coli to beta-lactam antibiotics. Figure 3-3: Ampicillin with Single Carbon Sources Has Limited Efficacy Against Stationary Phase E. coli. Survival of stationary phase cultures grown in 100% LB treated for 24 hours with 200 g/ml ampicillin (A, left) or 500 g/ml ampicillin (B, right) and 10 mM of the indicated carbon source. All individual replicates (6-7 for each treatment condition) are shown. NTC: no carbon control; Glc: glucose; Mann: mannitol; Fruc: frutose; Gly: glycerol; Gln: gluconate; Ara: arabinose; Xyl: xylose. Having found that single carbon sources can sensitize tolerant stationary phase E. coli to 1000 g/ml ampicillin, we next sought to determine if these carbon sources could sensitize lower concentrations of ampicillin. To do this, we took the carbon sources which showed efficacy in 100% LB and tested their ability to sensitize station- ary phase E. coli to a range of ampicillin concentrations. We found that one carbon source – xylose – was occasionally effective at 200 g/ml ampicillin (Figure 3-3A) 38
and most carbon sources showed occasional efficacy at 500 g/ml apmicillin (Figure 3-3B). These results are consistent with the failure of previous studies – which only tested a maximum concentration of 100 g/ml ampicillin – to observe sensitization of stationary phase E. coli to ampicillin by carbon source supplementation [2, 50]. As expected, metabolic counter-tolerance strategies are ampicillin-concentration- dependent (Figure 3-3). To understand the concentration-dependence of the carbon source stimulus, we next tested both 10 mM and 50 mM xylose. We found that 50 mM xylose sensitized lower ampicillin concentrations than 10 mM xylose (Figure 3-4). Additionally, at the ampicillin concentrations tested, the inter-replicate vari- Figure 3-4: Sensitization to Ampicillin by Xylose Depends Upon Xylose Concentration. Survival of stationary phase E. coli grown in 100% LB and treated for 24 hours with ampicillin and no xylose (NTC), 10 mM, or 50 mM xylose. Thick lines and symbols denote the average of two replicates. Thin lines and shaded area denote individual replicates and their range for each condition. ability observed with both 10 mM xylose and the other carbon sources (Figure 3-3) was not as pronounced with 50 mM xylose. Collectively, these results demonstrate that sensitizing tolerant E. coli to beta-lactam antibiotics is concentration-dependent in both the beta-lactam and the carbon source, and that previously tested conditions – such as 10 mM mannitol with 100 g/ml ampicillin – are just beyond the lower limit of effectiveness of this counter-tolerance strategy. Near this lower limit of effective- ness, inter-replicate variability leads to inconsistent results that mask the observation 39
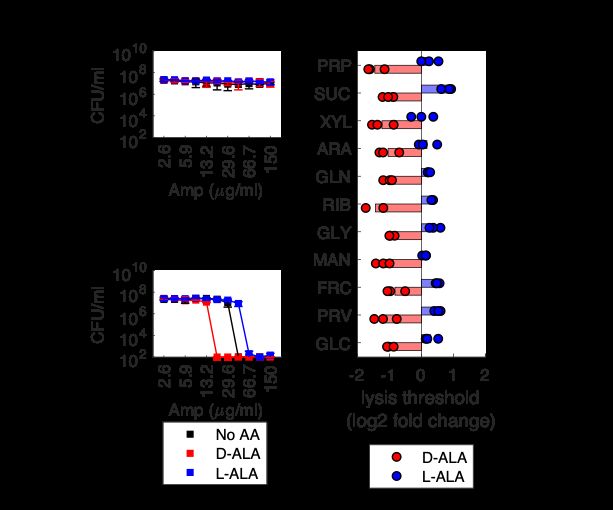
that supplementation with single carbon sources is indeed able to sensitize tolerant stationary phase E. coli to beta-lactam antibiotics. 3.4 Cell Growth May Contribute to but Does Not Explain Sensitization Next, we wanted to understand how cell growth contributes to sensitizing tolerant, stationary phase E. coli to ampicillin and other beta-lactams. It is commonly ac- cepted that beta-lactam lethality requires cell growth and that beta-lactam killing rates are directly proportional to bacterial growth rates [69, 119], though recent stud- ies have demonstrated that metabolic state and not growth rate determine beta- lactam lethality [79]. Therefore, we wanted to characterize cell growth in our exper- imental conditions to explore how cell growth contributes to restoring beta-lactam lethality. We first looked at 1% LB because at this low density we could rapidly quantify many different conditions using optical methods rather than CFU enumeration. For each carbon source, we tested a range of ampicillin concentrations and quantified the concentration at which lysis is observed (the lysis threshold) as well as any growth in the absence of ampicillin (Figure 3-5A). We would expect that if cell growth is key to beta-lactam sensitization that carbon sources that stimulate more growth would also stimulate lysis at lower ampicilin concentrations (Figure 3-5B). In contrast, we found a lack of correlation between cell growth and the metabolite-enabled ampicillin lysis threshold (Figure 3-5C). While these carbon sources did stimulate population-level growth as measured by biomass accumulation, the degree of biomass accumulation did not correlate with effectiveness in combination with ampicillin, suggesting that in 1% LB cell growth alone does not differentiate carbon soucres, and perhaps specific metabolic fluxes tune efficacy as was observed previously with aminoglycosides [87]. We next wanted to quantify cell growth in 100% LB. To do this, we sampled culture density at multiple time points during treatment and enumerated CFUs. We 40
You can also read

















































