NARBreakthroughArticle Mapping yeast mitotic 5 resection at base resolution reveals the sequence and positional dependence of nucleases invivo ...
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Nucleic Acids Research, 2021 1
https://doi.org/10.1093/nar/gkab597
NAR Breakthrough Article
Mapping yeast mitotic 5 resection at base resolution
reveals the sequence and positional dependence of
nucleases in vivo
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
Dominic Bazzano1,2 , Stephanie Lomonaco1 and Thomas E. Wilson 1,2,*
1
Department of Pathology, Ann Arbor, MI 48109, USA and 2 Department of Human Genetics, University of Michigan,
Ann Arbor, MI 48109, USA
Received May 09, 2021; Revised June 22, 2021; Editorial Decision June 23, 2021; Accepted June 28, 2021
ABSTRACT GRAPHICAL ABSTRACT
Resection of the 5 -terminated strand at DNA double-
strand breaks (DSBs) is the critical regulated step
in the transition to homologous recombination. Re-
cent studies have described a multi-step model of
DSB resection where endonucleolytic cleavage medi-
ated by Mre11 and Sae2 leads to further degradation
mediated by redundant pathways catalyzed by Exo1
and Sgs1/Dna2. These models have not been well
tested at mitotic DSBs in vivo because most methods
used to monitor resection cannot precisely map early
cleavage events. Here we report resection monitor-
ing with high-throughput sequencing using molec-
ular identifiers, allowing exact counting of cleaved
5 ends at base resolution. Mutant strains, including
exo1, mre11-H125N and exo1 sgs1, revealed a
major Mre11-dependent cleavage position 60–70 bp
from the DSB end whose exact position depended INTRODUCTION
on local sequence. They further revealed an Exo1- Accurate and efficient repair of DNA double-strand breaks
dependent pause point approximately 200 bp from (DSBs) is critical to cell survival and proper genomic func-
the DSB. Suppressing resection extension in exo1 tion (1). Two evolutionarily conserved pathways exist to re-
sgs1 yeast exposed a footprint of regions where pair DSBs: non-homologous end joining (NHEJ) and ho-
cleavage was restricted within 119 bp of the DSB. mologous recombination (HR) (2,3). In NHEJ, DSB ter-
These results provide detailed in vivo views of pre- mini are rapidly bound by the Ku heterodimer, a ring-like
vailing models of DSB resection and extend them to protein complex comprised of Yku70 and Yku80 in yeast
(4). Ku binding constrains nuclease activity at DSB ends
show the combined influence of sequence specificity
and promotes the association of downstream proteins crit-
and access restrictions on Mre11 and Exo1 nucle- ical to repair (5). The Mre11–Rad50–Xrs2 (MRX) com-
ases. plex also appears at DSBs nearly instantaneously and fur-
ther promotes downstream signaling, such as Tel1-mediated
checkpoint activation (6,7). MRX also has functions in
tethering the two DSB termini and end processing during
NHEJ (8,9). In NHEJ, the two DSB ends are ultimately lig-
ated by DNA ligase IV (Dnl4 in yeast) (10,11).
* To whom correspondence should be addressed. Tel: +1 734 764 2212; Email: wilsonte@umich.edu
C The Author(s) 2021. Published by Oxford University Press on behalf of Nucleic Acids Research.
This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial License
(http://creativecommons.org/licenses/by-nc/4.0/), which permits non-commercial re-use, distribution, and reproduction in any medium, provided the original work
is properly cited. For commercial re-use, please contact journals.permissions@oup.com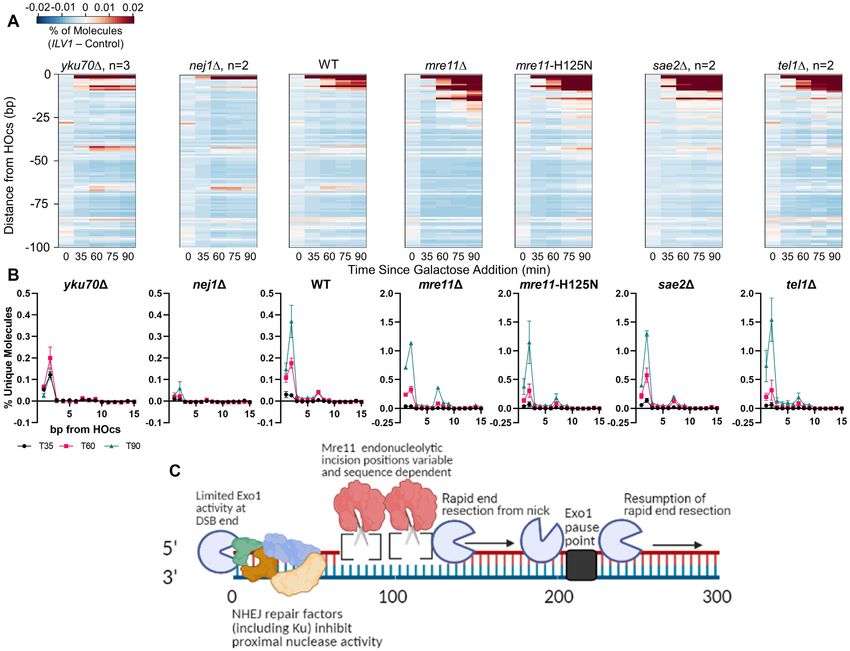
2 Nucleic Acids Research, 2021
Unlike NHEJ, HR relies on nucleolytic processing of gested a pause point in progressive resection. The pattern
DSB 5 -terminated strands in a mechanism called end re- in exo1 sgs1 mutant yeast exposed a ‘footprint’ of factors
section. This highly regulated process creates an exposed 3 - restricting cleavage activity near the DSB. Other mutants
terminated strand suitable for donor pairing and extension revealed dynamic activities at the extreme DSB terminus.
(12). End resection is proposed to begin through a targeted These studies provide a precise view of the nuclease activi-
endonucleolytic incision by Mre11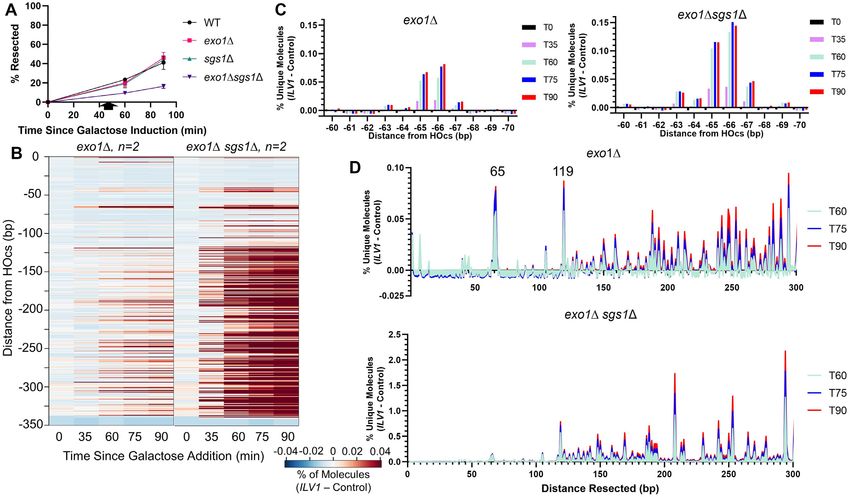
Nucleic Acids Research, 2021 3
saturation in YPA-dextrose media then inoculated to a low flanking the HOcs (FAM) and within an undamaged refer-
OD600 in YPA-glycerol for a consecutive night. When the ence gene, ACT1 (VIC). The fraction of uncut alleles was
OD600 of the YPA-glycerol cultures was in the range of directly measured as the fraction of molecules giving signal
0.3 to 0.6 to ensure exponential, asynchronous growth, the with the HOcs relative to the ACT1 probes. The true con-
strain with the control allele was spiked into the other YPA- trol allele spike-in percentage was further established in the
glycerol culture at a ratio of 1:20 as determined by OD600 . sequencing assay using DNA extracted from the T0 sample,
The T0 time point was taken by rapidly mixing 45 mL i.e., prior to DSB formation. That assay used paired probes
of this YPA-glycerol culture into 5 mL 0.5 M EDTA to homologous to the control allele (only present in the spiked-
quickly quench cellular nuclease activities. Galactose was in cells) and the HOcs-containing ILV1 locus (present in all
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
then added to the YPA-glycerol culture at 2% final concen- cells regardless of whether they had the control allele). Im-
tration to induce HO expression and DSB formation. An- portantly, all further time points were taken from the same
other time point was taken 35 min after galactose addition culture so the value from the T0 specimen applied to the
in the same fashion as T0. At 45 min after galactose addi- later time points also.
tion the YPA-glycerol + galactose culture was pelleted and
the cells resuspended in an equal volume of YPA-dextrose Restriction enzyme digestion, primer extension and in-
followed by continued shaking at 30 C. Further time points tramolecular ligation. Twenty micrograms of gDNA were
were taken as above at 60, 75 and 90 min after galactose digested by 40 units of NdeI (NEB) in Cutsmart Buffer at
addition (15, 30 and 45 min after the switch to dextrose, re- 37◦ C for 1 h. The tubes were then incubated at 65◦ C for 20
spectively). Time course materials used in ddPCR-based re- min to deactivate the NdeI. The digested DNA was puri-
section monitoring were generated by a similar protocol but fied using AMPure XP Beads and eluted using water in a
without the control allele spike-in and only 1.5 ml of cellu- volume of 60 l. The primer extension reaction used 1 unit
lar extract was taken from a smaller total growing culture of Phusion DNA polymerase in the provided buffer and a
volume without EDTA addition. custom primer containing a fixed 6 nt time point barcode
sequence, 12 nt hand mixed UMIs and several single fixed
nucleotides used to locate the UMIs (see below). Primer ex-
Genomic DNA preparation and quality validation
tension and tagging of molecules was done separately in
High molecular weight gDNA was obtained using the each time point with different fixed 6 bp barcode sequences.
Thermo Scientific Pierce Yeast DNA Extraction Kit. Yeast The uncycled extension reaction was: 98◦ C for 3 min, 55◦ C
cells were thus lysed without glass beads or damaging for 3 min, 72◦ C for 10 min. Twenty microliter of the eluate
chemical treatment. Genomic DNA was subjected to 0.8% following restriction enzyme digestion and purification was
agarose gel electrophoresis to confirm its high molecular added to three separate PCR tubes for each time point dur-
weight and purity (Supplementary Figure S3). Importantly, ing this step, with a final volume of 50 l in every reaction.
DNA is less prone to random fragmentation near the end of The extended molecules from each time point were purified
a DNA fragment, which was true for all stretches of DNA using AMPure XP and eluted using water in a final volume
we studied except for the ILV1-CR side of the DSB at time of 40 l. The entire eluate from each time point was diluted
0 before DSB formation. Those molecules placed the inter- into separate overnight intramolecular ligation reactions at
rogation region in the middle of a large RE fragment, which room temperature using T4 DNA Ligase (NEB) at a final
resulted in notably higher background as compared to the concentration of 6400 units in a 320 l reaction volume in
control allele at T0 (Figure 6A). This phenomenon resolved the provided buffer. The ligase was then inactivated at 65◦ C
by T35 because the HO DSB now placed the interrogation for 10 min.
region at the end of the source DNA molecule.
Amplification and sequencing of custom library. Once
UMIs had been covalently ligated to gDNA 5 endpoints
Resection monitoring by RE-ddPCR
and the ligase inactivated, all time point samples from a
To measure resection flux through the interrogation window given culture’s time course were pooled together and pu-
of our sequencing method, we used a ddPCR method based rified into 200 l water using AMPure XP beads. There-
on resistance of ssDNA to RE cleavage. That method is de- fore, any bias due to PCR amplification and sequencing
scribed in detail in Lomonaco et al. (30). Briefly, HinfI was was expected to apply equally to all time points. Specifi-
used to digest 300 ng gDNA from the smaller 1.5 ml cul- cally, ligation junctions were amplified with primers flank-
tures. HinfI cleavage was confirmed by gel electrophoresis. ing the HOcs that were tailed with sequences homologous
ddPCR measurements using probes targeting a HinfI site to Illumina P5 (DSB side) and P7 (upstream NdeI side)
355 bp from the HOcs and an ACT1 control locus revealed adapters used in high-throughput sequencing. This reaction
when the HinfI site became resistant to cleavage due to re- used KAPA HiFi HotStart Polymerase with the following
section. conditions: 95◦ C for 3 min, 17 cycles of 98◦ C for 20 s, 60◦ C
for 15 s, 72◦ C for 30 s, and finally 72◦ C for 1 min. Twenty
microliter of the ligated and purified eluate was added to
Resection monitoring by high resolution primer extension se-
10 separate PCR tubes, each a final volume of 50 l, for
quencing
every pooled strain time course during this amplification.
Quantification of DNA amount and the exact control allele Products were purified on AMPure XP beads and eluted in
ratio. gDNA was quantified and HOcs curves were estab- 125 l then a custom Illumina amplicon library was made
lished using a dual fluorophore ddPCR assay with primers through PCR using IDT Illumina-compatible i5 and i7 in-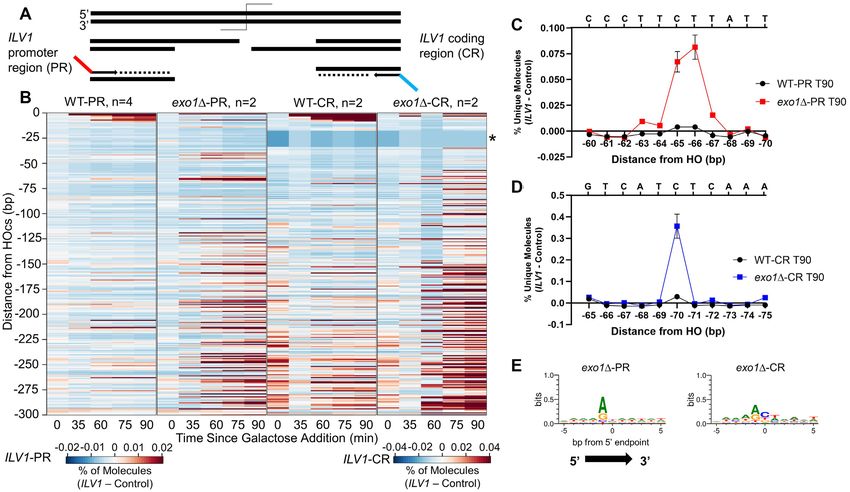
4 Nucleic Acids Research, 2021
dexed adapters (IDT10 UDI Adapter pairs #97 to #106), reference sequences and (iii) >95% of reads aligned with no
which identified the time course, i.e. source sample culture. mismatches.
The second adapter PCR also used KAPA HiFi with the fol-
lowing conditions: 95◦ C for 3 min and 8 cycles of 95◦ C for Purging redundant UMIs. We next accounted for PCR or
30 s, 55◦ C for 30 s, 72◦ C for 30 s. Fifty nanograms of the pu- sequencing errors introduced into UMIs that would lead
rified previous amplification product was put into this PCR to over-counting of the number of truly unique source
with a final volume of 50 l for each library. Ten individ- molecules. We used custom code to implement the direc-
ual custom amplicon libraries, each representing one com- tional model of Smith et al. (32), which considers a UMI
plete time course series, were submitted to the University of network to have originated from the UMI node with the
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
Michigan Advanced Genomics Core where they were bal- highest counts, with UMIs added to an existing network if
anced, pooled and sequenced on 2.5% of a shared Illumina they have at most one mismatch from a UMI already in the
Novaseq lane in the 2 × 150 format. network. Script ‘collapse/collapse UMIs.R’ implements an
efficient algorithm for solving such networks that matched
our needs, given that we expected very large numbers of
Bioinformatics analysis unique molecules mapping to a few specific alignment posi-
tions. The algorithm exploits 2-bit encoding of all possible
Code and file availability. All code and support files UMI sequences and pre-calculated XOR values for all UMI
required to execute data analysis are available from GitHub pairs with a Hamming distance of 1.
at https://github.com/wilsonte-umich/resection-seq. The Our 12 bp UMIs did not always have sufficient diversity
pipeline was executed using the q pipeline manager available to avoid UMI collisions for the HO or RE-cut ends of the
at https://github.com/wilsonte-umich/q-pipeline-manager. DSB allele; for this reason and for speed we disregarded
Briefly, a q data file establishes values for sample- and DSB allele positions that had >500K mapped molecules,
server-specific variables, which then invokes repository which always included the DSB end itself. In contrast, all
file ‘resection-seq/process sample.q’ via command ‘q positions were always subjected to UMI analysis for the
submit .q’. Each DSB end to be profiled control allele, which had been doped into the yeast pool
required an indexed FASTA file with the sequence of at a nominal fraction of 5% so that UMI collisions were
the reference and control alleles, plus Perl and R scripts minimal. The result was a reduction in molecule counts
defining their structures. Files for all DSBs used in per unique combination of time point, UMI sequence, and
this study are provided in repository directory ‘resection- alignment position to reflect only true source molecules.
seq/Bazzano 2021/references’. Each sample to be analyzed
required a row entry in a metadata file that described the Discarding failed libraries. To identify poorly
experiments and time points, provided as ‘resection- performant libraries we wrote repository script
seq/Bazzano 2021/ResectionMasterStrainTable.txt’. “resection-seq/parse q report.pl” to read the out-
put of ‘q report -j all .q’ and write a
Read validation and alignment. Data from the sequenc- table with summary statistics of all samples, pro-
ing core consisted of paired end reads that had been de- vided for all samples as repository file ‘resection-
multiplexed per sample using the Illumina indices but that seq/Bazzano 2021/ResectionOutputSummary.xlsx’. It
still had a mixture of all time points per sample. Read 2 allowed us to establish criteria that a time point li-
(from the P7 primer) was only needed for sample demul- brary needed to have at least 3.5M cell equivalents of
tiplexing and was not used further. Each read 1 from the P5 input DNA, as determined by the corrected control
primer was analyzed to determine if it matched, in read or- molecule count, and a background breakage frequency
der, (i) an appropriately long leader sequence corresponding less than 0.03% in the control allele. Libraries failing
to the DSB-distal end of the resected fragment, (ii) a UMI these criteria were written to repository file ‘resection-
of the appropriate length, (iii) a single fixed A base, (iv) a seq/Bazzano 2021/BadLibraryTable.txt’. Because time
known 6 bp time point barcode, (v) two fixed TA bases and point libraries were initially handled separately, a sample
finally (vi) sequence corresponding to the DSB-proximal or could have many successful time points even if one failed.
control-proximal DNA that had been ligated to the UMI
during circularization. Reads that failed to match this pat- Assembly, normalization and visualization. For each sam-
tern were discarded. ple we cross-tabulated read counts such that mapped posi-
For matching reads, 36 bp of the proximal sequence was tions were in rows and time points in columns and wrote
extracted and aligned to the DSB and control reference se- two R Shiny web tools to analyze these tables, provided in
quences using Bowtie (31) with options ‘-v 3 -k 1 -m 1 – repository directory ‘resection-seq/ server’; ‘visualize’ cre-
best’, such that only unique alignments with up to three mis- ates scatterplots for a series of samples as a function of allele
matches were accepted (some reads were expected to map position whereas ‘heatmap’ creates plots of position by time
twice to the common sequence shared between the DSB point with values expressed by color intensity.
and control alleles). Productive alignments were sorted and For each tool, the first normalization step was to sum
grouped by all unique combinations of time point, UMI se- the count of all unique control allele molecules, whether in-
quence, and alignment position, which acted as the defini- tact or fragmented, to establish the total number of con-
tive molecule identifier. Metrics consistently confirmed high trol alleles (and thus haploid cell equivalents) present in
data quality, including that (i) there were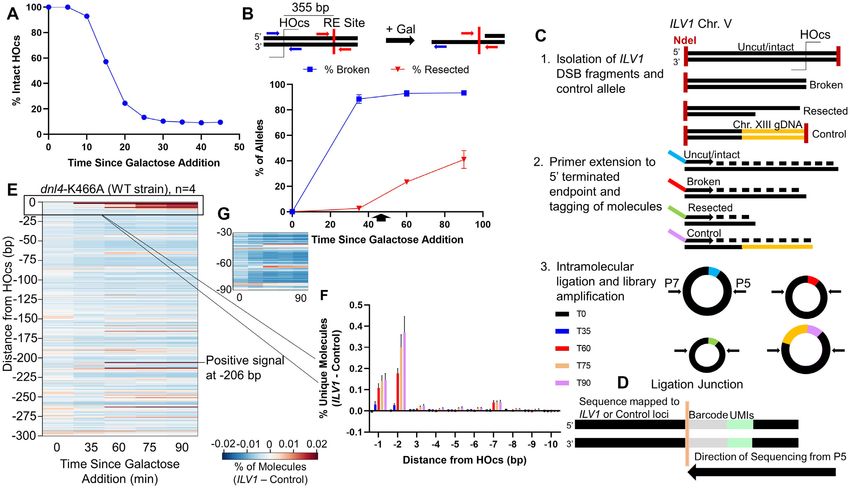
Nucleic Acids Research, 2021 5
recting that control allele count by the fraction of control to digestion by a restriction endonuclease (RE) (23). Care
allele-bearing cells in the sequenced mixture, determined was taken to extract high molecular weight genomic DNA
by ddPCR from the time 0 specimen as described above. (gDNA) given that sheared gDNA is indistinguishable from
The fraction of all input alleles that mapped to each pos- resection intermediates (Supplementary Figure S3).
sible DSB or control allele position was calculated by di- We further added a control allele to our strains that
viding the molecule count at that position by the actual or lacked a DSB but had (i) a shared common sequence with
inferred allele counts established above. Although extensive the DSB allele and (ii) a unique sequence abutting an NdeI
resection of the DSB allele could lead to lower total recov- restriction site, the same site as found 84 bp distal to the
ered counts at later time points, the calculated fractions re- ILV1 HOcs as well as within the shared common sequence
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
mained accurate because they were normalized to the con- (Figure 1C, panel 1, Supplementary Figure S1B, C). Puri-
trol allele, which was unaffected by DSB processing. fied gDNA was digested with NdeI followed by annealing
The signal for the control allele was mostly at the NdeI and extension of a primer that matched the NdeI site in
restriction site (Supplementary Figure S4) but the signal at the shared common sequence, which incorporated a unique
other control positions provided a means of assessing the molecular identifier (UMI) and a time point barcode into
extent and nature of random fragmentation that occurred in each gDNA molecule (Figure 1C, panel 2, Supplementary
each DNA prep. This pattern included evidence that smaller Figure S1B–D). Intramolecular ligation created a covalent
PCR fragments were sometimes more highly represented in bond between the UMIs and the 5 ends of both DSB and
final libraries, so we characterized the background fragmen- control DNA molecules, which might have been created
tation by fitting a linear regression to the non-NdeI posi- by NdeI, HO, 5 resection or random shearing (Figure 1C,
tions of the control allele. The fraction predicted by that re- panel 3). P5 and P7 sequences for high throughput sequenc-
gression was then subtracted from the fraction values at all ing were added by PCR amplification of pooled ligation
DSB positions to establish our final normalized values for products to minimize batch effects (Figure 1D). Because
DSB allele positions, referred to as ‘signal above control’ or the primer extension primer had a 5 hydroxyl, only gDNA
‘DSB – control’. Examination of samples at time 0 consis- strands with 5 phosphates could give final products.
tently validated that the control allele regression line was an After sequencing, unique source molecules were compu-
excellent model for background fragmentation of the DSB tationally assigned to the mappable positions of the DSB
allele (Supplementary Figure S4). and control alleles (see Methods). An average of 21M (range
Finally, normalized values were averaged for each time 8M to 34M) unique source molecules were obtained per
point over all samples of the same culture type (e.g. WT). time point (Supplementary File S1). We could not reliably
Heatmaps applied a linear color transformation to the av- quantify molecules matching the DSB HO and NdeI cleav-
eraged DSB signal above control; the scale of all plots is age positions due to UMI saturation but could quantify the
provided in the figures. The value can be negative due to control locus because we used a mixture in which only 5%
random fluctuations but is never expected to have large neg- of cells carried the control allele. Both loci had a minimal
ative values. background signal at internal positions before the DSB was
formed (Supplementary Figure S4). At 35 min, signal at the
Logo plots. Logo plots were generated using http:// DSB allele shifted to the HOcs position, referred to as posi-
weblogo.threeplusone.com/. ILV1-PR and CR % GC con- tion 0 (not shown), and at further time points we observed
tent (36 and 38%, respectively), were taken into account resection signal above background at positions internal to
when generating these plots. only the DSB allele (Supplementary Figure S4), referred to
with negative numbers to indicate how many bases had been
RESULTS removed from the HO cut end. For visualization, we sub-
tracted the control allele background signal from the DSB
Mapping end resection intermediates in NHEJ-deficient
allele signal and constructed heat maps (Figure 1E). Impor-
yeast
tantly, normalization to the control allele ensured that all
The DSB system was previously described and includes (i) signals reflect the absolute, not relative, fraction of DSB al-
the HO endonuclease coding sequence placed under con- leles that terminated at each position.
trol of the native GAL1 promoter and (ii) a single HO cut
site (HOcs) in a nucleosome-free region of the ILV1 pro-
Signal accumulations are evident at distinct positions even in
moter (Supplementary Figure S1A) (27,33). To facilitate re-
wild-type yeast
section monitoring without competing NHEJ, we added a
dnl4-K466A mutation to all strains that renders Dnl4 cat- Even though resection initiation through our ∼350 bp mea-
alytically inactive but able to bind normally to DSBs (29), surement window is expected to be efficient in our WT
henceforth referred to as wild-type (WT) with respect to strain (Figure 1B), we observed intriguing signal patterns
DSB resection. These alleles result in rapid and irreversible that are explored below. Molecules corresponding to the
generation of a site-specific DSB at IVL1 in >90% of cells first few base positions near the HOcs progressively in-
within 30 minutes of adding galactose to growing asyn- creased from 60 to 90 min after DSB induction (Figure
chronous yeast cultures (Figure 1A, Supplementary Figure 1E, F). Since the DSB had formed by 35 min, this increas-
S2). At 45 min after DSB induction, cultures were resus- ing signal at 60 min must be due to end processing asso-
pended in dextrose media to suppress HO expression and ciated with attempted NHEJ or resection initiation. There
stimulate resection (27,30) as measured in Figure 1B using a was also accumulation of signal only in the resecting time
ddPCR method based on the resistance of resected ssDNA points at a single −206 position (Figure 1E). Finally, when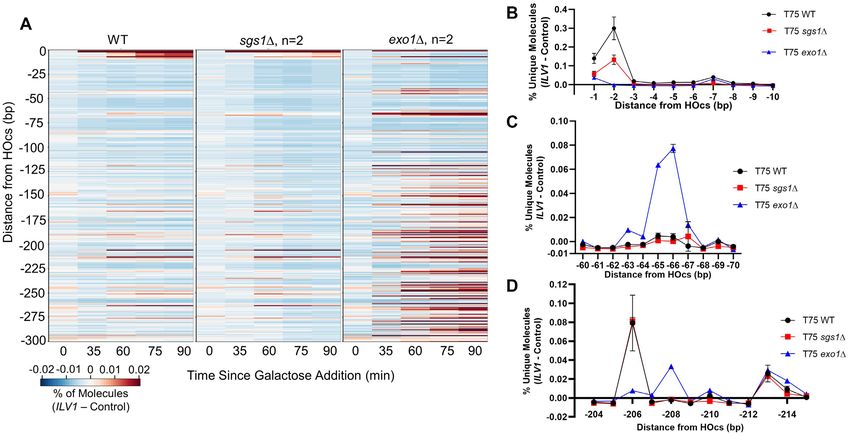
6 Nucleic Acids Research, 2021
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
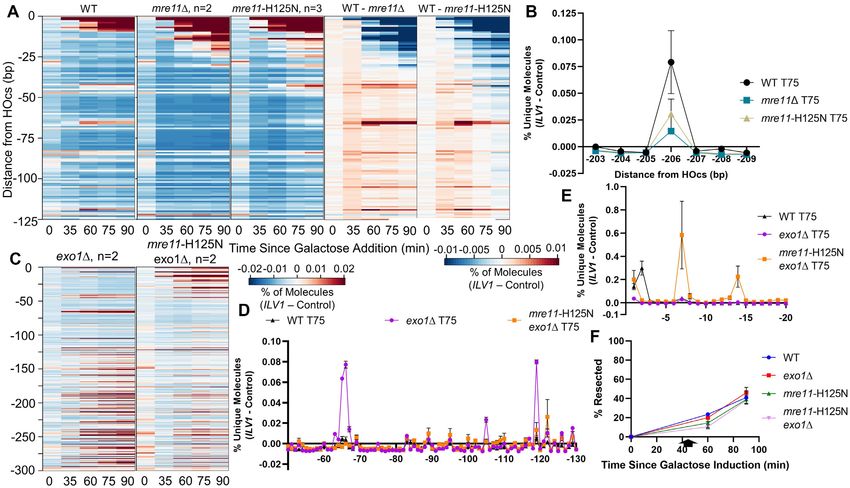
Figure 1. Description and validation of resection sequencing by primer extension and circularization. (A) ILV1 DSB formation efficiency as determined
by ddPCR after galactose addition at time 0 to induce HO expression. (B) ddPCR-based resection assay to determine the percentage of ssDNA after
DSB induction at a site 355 bp away from the HOcs. Cells were washed into glucose at 45 minutes (arrow). (C) Steps in resection sequencing to map 5
terminated end intermediates at base pair resolution using primer extension to add UMIs, followed by intramolecular circularization and PCR of junctions.
(D) Library products were sequenced such that an indexed P5 primer read, in order, the UMI, a fixed time point barcode and genomic bases from the
ILV1 DSB or a control locus. (E) Heatmap depicting the fraction of unique molecules with 5 endpoints at each position on one side of the ILV1 HOcs,
expressed as an absolute normalized signal above the background fraction of reads aligning to the control locus at similar fragment sizes (see Methods).
The NHEJ-defective Dnl4 catalytic point mutant dnl4-K466A is considered WT with respect to resection. (F) Bar graph showing the percentage of unique
ILV1 molecules with 5 ends within the first 10 bp away from the HOcs for each time point in WT. Negative position designations indicate the number of
bases removed from the 5 terminated strand. Bars are the average ± standard deviation of four biological replicates. (G) Expansion of the region −30 to
−90 bp from the HOcs in WT. The color scale is 2-fold more sensitive than (E).
we looked near the −30 and −60 positions suggested as pressing it in WT cells. There was minimal signal in exo1
Mre11 cleavage positions by the biochemical experiments after the −65/−66 bp position until −119 bp from the DSB,
of Cejka and Sung, we noted a slight increase in signal at after which we observed a steady increase in intermediates
selected positions, especially at −65/−66 (Figure 1E). This throughout the sequencing window. Despite this general in-
signal was weak but compelling due to its location, similar crease in reads mapping further away from the DSB termi-
time course as above, and the negative footprint surround- nus in exo1, loss of Exo1 led to a specific reduction in
ing it where little cleavage was observed (Figure 1G). read accumulation at the -206 bp position, identifying it as a
likely pause point for progressive Exo1-mediated resection
(Figure 2A, D). The large difference when comparing sgs1Δ
Exo1 loss shifts the DSB end resection pattern more than the and exo1Δ resection patterns suggests that Exo1-mediated
loss of Sgs1 resection is the preferred mode of end resection initiation
To add information to the signal patterns above we repeated near the DSB. Importantly, ILV1 read accumulations were
resection sequencing in strains lacking Exo1 or Sgs1, which nearly identical in independent biological replicates (Sup-
are required for the two main long-range resection mech- plementary Figure S5).
anisms (16,34). While the sgs1 resection profile looked
largely like that of WT (Figure 2A), exo1 first led to a
Mre11 is responsible for the peaks at the −65/−66 and other
decrease in reads aligning near the HOcs, especially at the
positions
−2 position, consistent with Exo1 itself being responsible
for removing a limited number of terminal bases (Figure Results above demonstrate that a protein other than Exo1
2A-B) (35). In marked contrast, we observed a dramatic in- must create the signal peak at the −65/−66 position, which
crease in reads mapping to the -65/-66 bp position in the prevailing models predict should be Mre11. To explore
absence of Exo1 (Figure 2A, C), indicating that Exo1 did this, we first examined strains with a complete deletion of
not create this signal but instead was responsible for sup- MRE11. Sensitive examination surrounding the −65/−66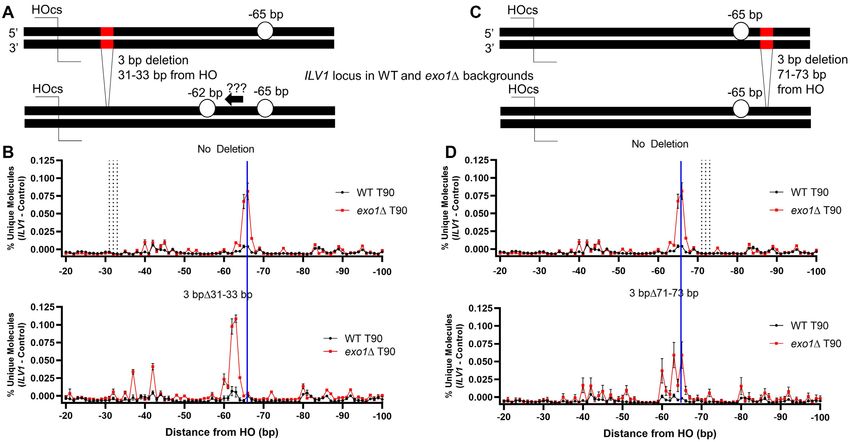
Nucleic Acids Research, 2021 7
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
Figure 2. Loss of Exo1 increases resection signal close to the ILV1 DSB, especially at -65/-66 bp. (A) Heatmaps of resection signal similar to Figure 1E for
WT, exo1 and sgs1 yeast. (B−D) The normalized fractional resection signal above background is plotted for WT, sgs1 and exo1 yeast at positions
(B) −1 to −10 bp, (C) −60 to −70 bp and (D) −204 to −215 bp from the ILV1 HOcs on the promoter side of the gene. Exo1 loss had opposite effects on
the signal obtained at the −2 and −206 positions as compared to the −65/−66 bp peak.
position similar to Figure 1G revealed that loss of Mre11 minutes and then decreased whereas it remained persistent
erased the small signal peak observed in WT cells, although in exo1 mutant strains (Supplementary Figure S6), consis-
intriguingly the negative footprint surrounding that posi- tent with a kinetic model in which Exo1 acts later at the ac-
tion remained even without Mre11 present (Figure 3A). tivated nick created by Mre11. The −119 position appears
With the benefit of Mre11 loss we could also discern that to be a major transition point as reads are suppressed in
less intense signal peaks were present in WT at −42, −83, the region between −65 and −119 and increased after −119
−105 and −119 (Figure 3A, Supplementary Figure S6). through the end of our sequencing window in exo1 mutant
Here, it is important to note that our method can only yeast.
map a single 5 terminal position of a source molecule rela- In contrast, we observed a marked increase in interme-
tive to the DSB-distal primer. Thus, we cannot definitively diates mapping near the HOcs in the mre11-H125N exo1
judge how effectively the more DSB-proximal −42 posi- double mutant (Figure 3C, E). Resection mediated by Sgs1-
tion is cleaved relative to the efficient −65/−66 position, Dna2 is the primary pathway in the absence of both Mre11
since cleavage at −65/−66 would prevent detection of si- nuclease activity and Exo1 (16). Therefore, the increase in
multaneous cleavage at −42. We can confidently state that reads near the DSB end in mre11-H125N exo1 might re-
the −65/−66 position is cleaved more efficiently than the flect a shift to resection carried out from the end by Sgs1-
more DSB-distal positions. Importantly, mre11 also re- Dna2 as opposed to resection beginning from the nick cre-
duced most signal in our interrogation window after the first ated by Mre11. Intriguingly, analysis of resection kinetics
∼10 bp, including the −206 peak (Figure 3B). through our sequencing window via the RE-ddPCR as-
To establish the actions of Mre11 more definitively we say showed only a small resection defect in mre11-H125N
added the mre11-H125N mutation, which abolishes the exo1 double mutant yeast compared to mre11-H125N
Mre11 nuclease activity (36), to our WT and exo1 strains. and exo1 single mutants alone (Figure 3F). There is strong
The mre11-H125N strain by itself behaved similarly to evidence that Mre11 has end-chewing activity that aids in
mre11 implicating the Mre11 nuclease as being required Sgs1 loading (37), but the presence of these intermediates
for the increase in distal resection intermediates (Fig- in mre11-H125N exo1 indicates that another protein also
ure 3A). Examination of mre11-H125N exo1 relative to has such activity.
exo1 revealed a complete loss of signal at −65/−66 and
substantially reduced signal at positions −119 and beyond
Combined loss of Sgs1 and Exo1 reveals the profile of Mre11
(Figure 3C, D). These results expose an additional stepwise
activity permissible at a DSB
cleavage event mediated by Mre11 at position −119, 53 bp
from position −66. Notably, at both the −65/−66 and −119 It is known that Mre11 is responsible for ∼300 bp of re-
positions the signal peaked in EXO1 wild-type strains at 60 section in exo1 sgs1 double mutant yeast defective in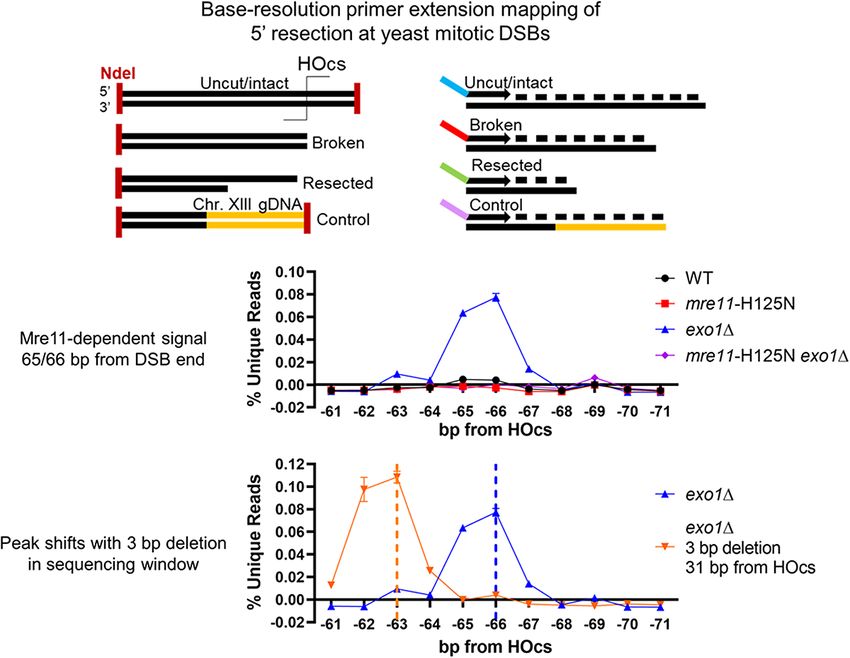
8 Nucleic Acids Research, 2021
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
Figure 3. Mre11 nuclease activity is necessary to form resection intermediates at −65/−66 bp. (A) The first three panels show heatmaps of resection signal
for WT, mre11 and catalytically defective mre11-H125N strains in the first 125 bp from the HOcs. The next two panels plot the difference between the WT
and mutant strains to highlight where signal was higher (red) or lower (blue) in WT as compared to mutant. (B) Fractional resection signal at positions
−203 to −209 bp in WT, mre11 and mre11-H125N strains at the 75-min time point. (C) Heatmaps of resection signal for exo1 and mre11-H125N
exo1 yeast up to 300 bp from the HOcs. (D, E) Fractional resection signal 75 minutes after DSB induction in WT, exo1 and mre11-H125N exo1 yeast
at positions (D) −50 to −130 bp and (E) −1 to −20 bp from the HOcs. (F) ddPCR-based resection analysis of WT and mutant strains at a site 355 bp from
the HOcs. Points are the average of two biological replicates.
long range resection (38). Indeed, ddPCR quantification proximal to this point, the presumptive actions of Mre11 are
of ssDNA produced from resection in exo1 sgs1 yeast restricted to only a few selected positions that have an im-
showed a decrease in resection efficiency in this strain as perfect periodicity (Supplementary Figure S6). After −119
compared to WT (Figure 4A). Analysis of exo1 sgs1 bp, the resection pattern shifts in both exo1 sgs1Δ and
yeast in our sequencing assay revealed an increase in signal exo1Δ to one indicative of a kinetically slower process, since
at similar positions as observed in the single exo1Δ mutant once again cleavage at the more distal positions would pre-
strain (Figure 4B,D, note the scale difference in Figure 4D). vent detection of simultaneous cleavage at the more prox-
The only locations where reads were decreased in exo1 imal positions. It is noteworthy that major peaks occur at
sgs1 compared to exo1 or sgs1 single mutants were at positions −208 and −294 in the double mutant, which may
the positions immediately proximal to the DSB termini, in- represent further stepwise incisions generated by Mre11.
dicating that end chewing events in resection are dependent
on the presence of Sgs1 or Exo1.
Mre11 endonucleolytic activity is influenced by DNA se-
Interestingly, the percentage of unique reads at the -65/-
quence preferences
66 bp position was only modestly elevated in exo1 sgs1
as compared to exo1 (Figure 4C), which contrasts with the Broadly, three mechanisms might dictate the specific resec-
large (>10-fold) increase in reads throughout the sequenc- tion profiles seen above: (i) proteins might ‘measure’ cleav-
ing window further from the DSB in the double mutant age positions relative to the DSB end, (ii) pre-damage fac-
(Figure 4B,D). We observed intermediates mapping to po- tors such as nucleosomes or other bound proteins might
sitions throughout our sequencing window without a drop constrain repair protein action, or (iii) repair proteins them-
off in signal, which indicates that Mre11 or another pro- selves might show a driving sequence dependence. To dis-
tein is resecting past the positions that can be mapped by tinguish these in our system, we made two deletions near
the assay (344 bp), even when we extended our window the −65/−66 position in WT and exo1 backgrounds (Fig-
to include reads mapped to the common sequence shared ure 5A, C). The deletions each removed three base pairs
with the control allele (Supplementary Figure S7). The mas- either 31–33 or 71–73 bp away from the HOcs, i.e. DSB
sive increase in signal in exo1 sgs1 clearly exposed the proximal and distal to the −65/−66 position, respectively
transition point at position −119 (Figure 4B, D). DSB- (Supplementary Figure S1B). Strikingly, the DSB-proximalNucleic Acids Research, 2021 9
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
Figure 4. Exo1- and Sgs1-deficient yeast show greatly enhanced resection intermediates beyond position −119. (A) ddPCR-based resection analysis of WT
and mutant strains at a site 355 bp from the HOcs with combinations of exo1 and sgs1 deletions. (B) Heatmaps of resection signal for exo1 and exo1
sgs1 yeast. The uniquely mappable ILV1 sequences end at position -344. (C) Fractional resection signal surrounding the −65/−66 bp position for exo1
and exo1 sgs1 yeast. The same y-axis scaling is used on both graphs. (D) Fractional resection signal for the first 300 bp away from the DSB for each
time point in exo1 and exo1 sgs1 yeast. Note the large difference in y-axis scaling due to the accumulation of many more intermediates in the exo1
sgs1 strain.
deletion led to a −3 bp shift in the position of the Mre11- ther side of the break (Figure 6B). Intriguingly, a number
dependent peak in both WT and exo1 cells, meaning that of reads lined up at the ILV1-CR −206 bp position in both
the same underlying sequence was being cleaved by the WT and exo1 cells. Of primary note was that a peak ap-
Mre11 nuclease even though it was now closer to the DSB peared at −70 bp for ILV1-CR and not at −65/−66 bp as
end (Figure 5B). Somewhat unexpectedly, the deletion dis- for ILV1-PR, consistent with the sequence, not distance, de-
tal to the −65/−66 position changed the pattern of peaks pendence noted above. Analysis of the underlying sequence,
at positions −60 to −66, likely due to their proximity to oriented similarly with respect to the DSB, revealed a simi-
the deletion (Figure 5D). Both deletions further shifted the lar palindromic ‘TCT’ motif, with the cytosine being an ef-
resection pattern throughout the distal portions of the se- ficient incision point (Figure 6C, D). Examination of the re-
quencing window, including the Exo1-dependent -206 bp gion around −119 in ILV1-CR exo1 mutants revealed read
position in WT cells and the −119 position in exo1 (Sup- accumulations at −125 and −153, 55 and 83 bp after the
plementary Figure S8). Thus, the signal patterns through- −70 position, respectively.
out the interrogation window proved to be highly sequence, To examine the nucleotides contributing to the exo1-
not distance, dependent. dependent rise in resection signal in both sequencing win-
dows, we first set a cut-off value of 10% or 20% of the high-
est number of unique reads counted at one position in each
Sequencing of 5 endpoints formed from both ends of the same
exo1 mutant. We then generated logo plots of the DNA se-
DSB allele
quence surrounding these 5 terminated endpoints (Figure
To expand our base of observation we next examined the 6E, Supplementary Figure S9). We observed a slight pref-
opposite side of the ILV1 DSB using the same conceptual erence for A or G in the position immediately before the
approach as in Figure 1 (Supplementary Figure S1A). Be- 5 -terminated endpoint in Exo1-deficient cells. Even though
cause this second side of the DSB corresponds to the ILV1 this sequence-specific signal was weak, it is noteworthy be-
coding region, we refer to it as ILV1-CR and the sequence cause it is different than the TCT sequence noted above.
studied above as ILV1-PR, for ‘promoter’ (Figure 6A). We However, a C preference on just the ILV1-CR side of the
observed a higher background in WT and exo1 cells for DSB became apparent for the more restricted set of peaks
ILV1-CR, but a similar overall pattern was observed on ei- at the 20% threshold (Supplementary Figure S9).10 Nucleic Acids Research, 2021
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
Figure 5. The predominant Mre11-dependent incision shows sequence dependence more than a strict positional dependence. (A) Diagram of a 3 bp deletion
in ILV1 located 31–33 bp from the HOcs. (B) Fractional resection signal from positions −20 to −100 in WT and exo1 yeast 90 min after DSB induction
with and without the 3 bp deletion at positions −31 to −33. Vertical dashed lines in the top plot mark the location of the positions that were deleted in
the bottom plot. A vertical blue line indicates the position expected for the peak in the bottom plot if the distance from the DSB end had been maintained
after the 3 bp deletion. (C) Diagram of a 3 bp deletion in ILV1 located 71–73 bp from the HOcs. (D) Similar to (B) for the 3 bp deletion located at positions
−71 to −73.
NHEJ and other repair factors influence resection initiation DISCUSSION
near the DSB terminus
End resection is a fast process occurring at a rate of 4
To gain further insight into the obstacles that Mre11 faces kb/h (34). However, HR must overcome multiple obsta-
near the DSB terminus, we deleted the NHEJ genes YKU70 cles close to the DSB that dictate the successful transition
and NEJ1 and analyzed early resection sequencing inter- to long-range resection (39). One such obstacle is repair by
mediates. Intriguingly, the signal at position −65/−66 per- NHEJ. We exploited catalytically defective Dnl4 to remove
sisted in yku70, indicating that Ku binding to a DSB end is this competing outcome while maintaining normal Ku and
not uniquely responsible for this Mre11-dependent cleavage DNA ligase IV assembly at DSB ends (29). Despite these
position (Figure 7A). However, the more proximal −42 bp obstacles, >20% of HO-cut ILV1 alleles were resected past
peak became more prominent in yku70. Moreover, loss of our ∼350 bp sequencing window within 60 min of suffering
Ku created an interesting change near the DSB wherein WT the DSB in asynchronous yeast cultures. Resection steadily
showed a progressive increase in signal over time whereas increased to ∼40% of alleles at 90 minutes but was asso-
yku70 had a nearly constant signal (Figure 7B), which ciated with only limited detectable 5 -endpoint patterns in
might suggest an easier movement of resection inward from strains that were wild-type for resection-associated genes.
the DSB end. More drastic was the decrease in reads align- Thus, while resection was clearly initiating in the sequenced
ing near the HOcs in nej1, which may also signify an regions during the experiment, the transition to progressive
increase in the kinetics of resection initiation. Restriction- long-range resection was normally kinetically very fast such
based ddPCR measurements of end resection kinetics con- that few endpoint detections accumulated in our observa-
firmed a higher occurrence of ssDNA at a site 355 bp from tion window.
the HOcs in these mutants compared to WT (Supplemen- Mutations further revealed important features of the ob-
tary Figure S10). served patterns, summarized in Figure 7C, where interpre-
Finally, examination of factors involved in supporting tations are guided by the following principles. When a sig-
cleavage by Mre11 revealed an increase in reads aligning nal peak is observed, we infer that there is a relative delay
near the HOcs in sae2 and tel1 (note the scale differ- (not necessarily a block) in processing leading to increased
ence between WT and these mutants in Figure 7B) and the steady-state signal for those intermediates. When a strain
same near absence of reads at −65/−66 bp as in mre11Δ alteration causes such a signal to increase then that delay
mutants. These results confirm a slower 5 degradation pro- has been accentuated, which implicates the altered protein
cess from the DSB end in the absence of Mre11 nuclease as being responsible for normal processing away from that
activity, specifically at the Exo1-dependent −2 position. 5 -terminated intermediate. In contrast, decreased signal inNucleic Acids Research, 2021 11
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
Figure 6. Comparing two sides of the ILV1 DSB reinforces sequence dependence more than a strict positional dependence. (A) Sequencing scheme to
capture intermediates resecting into the ILV1 coding region (ILV1-CR; Figures 1–5 examined resection into the ILV1 promoter region, ILV1-PR). See
annotated map and sequences in Supplementary Figure S1. (B) Heatmaps of resection signal up to 300 bp from the HOcs in WT and exo1 strains
sequenced from either ILV1-PR or ILV1-CR in separate libraries. The scale for ILV1-CR strains is 2-fold less sensitive due to higher background levels.
The ILV1-CR region marked with an asterisk failed to return any aligned reads for technical reasons due to an adjacent T-rich stretch of bases. (C)
Fractional resection signal at positions -60 to −70 bp in WT and exo1 yeast sequenced from ILV1-PR. The sequence of the 5 terminated strand is shown
on the upper X-axis. (D) Fractional resection signal at positions −65 to −75 in WT and exo1 yeast sequenced from the ILV1-CR, with the sequence
as in (C). (E) Logo plot analyzing sequences ±5 bp in either direction from signal peaks in exo1 yeast sequenced from either ILV1-PR or ILV1-CR. A
threshold of 10% of the max peak height was used to determine the plotted peaks: ILV1-PR n = 75, ILV1-CR n = 67. See Supplementary Figure S9 for
depiction of these thresholds relative to peak intensities.
response to a strain alteration suggests that the protein fac- mediates. Notably, signal at the -2 position was uniquely
tor normally creates the signal; such an intermediate can dependent on Exo1, which identifies Exo1 as the nuclease
sometimes be inferred to be the product of a nuclease’s ac- that cleaves some, but not all, of the DSB-proximal 5 inter-
tion. However, a caveat is that primer extension only reveals mediates (42). In contrast to Ku and Nej1, loss of Mre11
the most DSB-distal 5 cleavage point on a DNA strand, or its nuclease activity led to increased signal at the DSB-
so that DSB-proximal increases could result from decreased proximal positions, most profoundly at −2 bp, with a simi-
endonucleolytic activity more distally and vice versa. lar result upon loss of the Tel1 and Sae2 proteins that sup-
Unlike our base dnl4-K466A mutation, yku70 and nej1 port Mre11 (7,13,43). Here, the best interpretation is that
mutations removed the Ku and Nej1 NHEJ factors thought the signal increase is secondary to decreased DSB-distal en-
to be inhibitory to resection (14,40). The greatest effect of donucleolytic cleavage.
these losses was indeed to decrease the signal near the DSB. Moving inward from the DSB end we encounter the most
Thus, Ku and Nej1 might normally impede further pro- critical region for resection initiation. Previous in vitro anal-
cessing away from these positions. Loss of Nej1 had the ysis of Mre11 activity on Ku-occluded dsDNA substrates
largest impact and reduced signals to near-background lev- revealed incisions at 35–45 and 55–65 bp from the end on
els, consistent with a function of Nej1 in limiting resection 70 and 100 bp substrates, respectively (19,20), leading to
away from the DSB and thus promoting endpoint detec- the prevailing model wherein Mre11 creates a nick at a dis-
tions near the DSB (41). An alternative interpretation is that tance enforced by the Ku protein block (44). We observed
the DSB-proximal signals are not resection intermediates a pattern of iterative peaks throughout this region con-
but instead reflect processing associated with futile attempts sistent with the model and with the inference of a ‘step-
at NHEJ. These are not exclusive concepts and the fact that wise’ cleavage at somewhat, although not precisely, regu-
signal at the prominent -2 position failed to accumulate in larly spaced positions (21). Although subtle, the pattern of
Ku mutant yeast suggests that NHEJ factors might nor- multiple ILV1-PR peaks at −42, −65/−66, −83, −105 and
mally, but only temporarily, stabilize DSB-proximal inter- −119 bp positions were evident in wild-type yeast and en-12 Nucleic Acids Research, 2021
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
Figure 7. NHEJ factors limit processing events near the DSB end (A) Heatmaps of resection signal within 100 bp of the ILV1-PR HOcs in WT and various
single deletion mutants. (B) Fractional resection signal within the first 15 bp after the ILV1-PR HOcs for the same mutants as in (A). (C) Model for the
initiation of mitotic end resection in yeast that expands on prevailing bi-directional and stepwise models as informed by single base resolution resection
monitoring. See text for discussion. Model figure created using BioRender.
hanced upon loss of Exo1. That increase was strongly de- sion position at −70 bp on the ILV1-CR side of the DSB
pendent on the Mre11 nuclease as evidenced by absence of whose sequence shared 5 out of 7 bases with the ILV1-PR
−65/−66 and −119 signal in an exo1 mre11-H125N dou- −65/−66 peak. Critically, this predominant cleavage posi-
ble mutant. The relative weaknesses of the −42 signal could tion did not move in yku70 mutant yeast but the −42 po-
be secondary to cleavage at −65/−66, but −83 had substan- sition did become the most prominent. Thus, Ku itself is
tially less activity, establishing that the different cleavage po- apparently not the primary factor leading to the position of
sitions are utilized with different efficiencies. the inferred Mre11 incisions but it may impede access to the
A central question is what determines the pattern of more DSB-proximal positions.
cleavage at mitotic DSBs in vivo. Models cited above im- It is equally important to consider where resection in-
ply that Ku and/or Mre11 protein sizes are critical factors termediates were not observed, which was especially no-
(19,20,44), but the pattern could be enforced by other pro- ticeable in exo1 sgs1, a strain deficient in long range re-
teins bound prior to damage such as histones or other pro- section thought to mainly support cleavage by Mre11 (38).
teins (45,46) and DNA repair proteins can have sequence- Resection initiation signal with this strain increased dra-
specific properties (47,48). We addressed these issues using 3 matically, although the signal at −65/−66 was only mod-
bp deletions in the ILV1-PR sequence. Strikingly, the -65/- estly increased relative to exo1. Indeed, most positions up
66 peak moved closer to the DSB with the DSB-proximal to −119 bp had limited read counts despite the poor ability
deletion and thus tracked the local sequence, not the dis- to transition to progressive resection. We infer that the first
tance from the DSB end. Further evidence for sequence ∼120 bp of a DSB are a unique zone where a specific pro-
specificity comes from the identification of a similar inci- tein ‘footprint’ protects the DNA from promiscuous activ-Nucleic Acids Research, 2021 13
ity and where Mre11-dependent cleavage is preferred. The that phenomenon corresponds to our precise Exo1 pause
source of that protection is not entirely clear, but it is not point (24).
Ku, Sgs1, or Exo1 based on our data. It might be Mre11- In summary, results here provide a uniquely high-
Rad50 itself, although this is difficult to prove with resection resolution picture of mitotic DSB resection initiation. They
data alone. support published models in which Ku and Nej1 inhibit ex-
The genesis of signal after −119 bp is intriguing. In yeast onucleolytic resection from the DSB end in a manner that
with Exo1 and Sgs1 it is dependent on the Mre11 nuclease. is overcome by Mre11 incision with a peak activity ∼60 to
This is especially true for the strong and specific -206 posi- 70 bp from the DSB end but with evidence for multiple in-
tion, which we believe results from exonucleolytic extension cision points that might occur in a stepwise fashion. Subse-
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
away from the more proximal incisions. Consistently, the - quent nick extension by predominantly Exo1 is rapid and
206 signal was abrogated in exo1 yeast suggesting it as a processive but also subject to pausing (Figure 7C). How-
pause point during extension by Exo1. We do not know the ever, the cleavage pattern is complex, substantially sequence
nature of the obstacle that causes Exo1 to pause but one dependent, and influenced by an inferred protective protein
candidate is the 9–1–1 complex based on recent observa- complex that is not strongly dependent on Ku. Critical chal-
tions (38). Whatever is responsible is again sequence specific lenges are to extend these findings across many DSBs and to
as the location of the peak shifted with the DSB-proximal correlate DNA-based results with equally high-resolution
3 bp deletions. The signal accumulation at nearly every po- protein binding studies in vivo to understand the nature of
sition past -119 bp in exo1 sgs1 yeast is uncertain; it might the early mitotic repair protein complex.
be a result of more distal, kinetically slower and pathologic
cleavage by Mre11 but we were unable to make an exo1 sgs1
DATA AVAILABILITY
mre11-H125N triple mutant to test this idea (16).
A caveat is the possibility for artifacts arising from the FASTQ files with sequencing reads are available for all
use of the HO endonuclease. HO can remain bound to its samples from the NIH Sequence Read Archive via acces-
cleavage product (49) so we cannot rule out that HO itself sion PRJNA703820. Code and additional summary pro-
is partially responsible for the observed cleavage patterns, cessed data files are available on GitHub: https://github.
although all DSB repair actions occur at HO DSBs and com/wilsonte-umich/resection-seq.
we make our measurements long after repair proteins have
engaged the DSB. We also considered whether HO might
cleave off-target to create some measured endpoints, espe- SUPPLEMENTARY DATA
cially when the DSB was resistant to resection (50). One Supplementary Data are available at NAR Online.
peak at –294 in exo1 sgs1 yeast had possible sequence simi-
larity to the HOcs and could represent ‘skipping’ of HO to
this alternative position (Supplementary Figure S11). Our ACKNOWLEDGEMENTS
studies also have a limited ability to determine the extent The authors thank Olivia Koues and the staff of the Michi-
to which nucleosomes might help drive resection cleavage gan Advanced Genomics Core for advice and expert han-
positions, in part because we used a DSB in a nucleosome- dling of high throughput sequencing services. We also thank
free region of a gene promoter, which may be less generaliz- Christian Rizza for his work assembling the RE-ddPCR-
able to other chromatin (33). Such a promoter region is not based resection plots and former lab members Dongliang
necessarily devoid of all nucleosome binding, including by Wu, James Daley and others for input and discussions over
fragile nucleosomes and nucleosome variants (51), while at the course of developing this project.
the same time the DSB repair response is known to engage
in nucleosome repositioning (52). Finally, because we used
asynchronous cultures our results represent an average of all FUNDING
cell cycle stages. We performed DNA content analysis of a National Institutes of Health [GM120767 to T.E.W., and a
subset of our strains in a parallel study, which showed some supplement of the same number to S.L.]. Funding for open
G1 resection even in WT and that even exo1 sgs1 yeast have access charge: National Institutes of Health [GM120767 to
some arrest in response to the unrepaired DSB (30). Dif- T.E.W.].
ferences in cell cycle distributions in mutants thus do not Conflict of interest statement. None declared.
appear sufficient to account for the large changes in base-
resolved resection patterns, which are much more consistent
with a direct mechanistic action of proteins known to bind REFERENCES
DSBs. 1. Scully,R., Panday,A., Elango,R. and Willis,N.A. (2019) DNA
Our results align with studies that used sequencing ap- double-strand break repair-pathway choice in somatic mammalian
proaches to study meiotic end resection. Notably, we ob- cells. Nature reviews. Mol. Cell Biol., 20, 698–714.
2. Chapman,J.R., Taylor,M.R. and Boulton,S.J. (2012) Playing the end
served increased intermediates throughout our sequencing game: DNA double-strand break repair pathway choice. Mol. Cell,
window in exo1 cells that mirrors the effect reported by the 47, 497–510.
Keeney group (24,25). Our data also support the observa- 3. Zhao,B., Rothenberg,E., Ramsden,D.A. and Lieber,M.R. (2020) The
tions that Sae2 and Tel1 promote efficient resection and a molecular basis and disease relevance of non-homologous DNA end
joining. Nat. Rev. Mol. Cell Biol., 21, 765–781.
decrease in unique reads mapping near the DSB termini 4. Emerson,C.H. and Bertuch,A.A. (2016) Consider the workhorse:
(24,25,53). Keeney described evidence of Exo1-dependent Nonhomologous end-joining in budding yeast. Biochem. Cell Biol.,
pausing at heterochromatin but it is uncertain how closely 94, 396–406.14 Nucleic Acids Research, 2021
5. Frit,P., Ropars,V., Modesti,M., Charbonnier,J.B. and Calsou,P. double-strand break in Saccharomyces cerevisiae. Genetics, 178,
(2019) Plugged into the Ku-DNA hub: The NHEJ network. Prog. 1237–1249.
Biophys. Mol. Biol., 147, 62–76. 28. Palmbos,P.L., Daley,J.M. and Wilson,T.E. (2005) Mutations of the
6. Myler,L.R., Gallardo,I.F., Soniat,M.M., Deshpande,R.A., Yku80 C terminus and Xrs2 FHA domain specifically block yeast
Gonzalez,X.B., Kim,Y., Paull,T.T. and Finkelstein,I.J. (2017) nonhomologous end joining. Mol. Cell. Biol., 25, 10782–10790.
Single-molecule imaging reveals how Mre11-Rad50-Nbs1 initiates 29. Chiruvella,K.K., Liang,Z., Birkeland,S.R., Basrur,V. and
DNA break repair. Mol. Cell, 67, 891–898. Wilson,T.E. (2013) Saccharomyces cerevisiae DNA ligase IV supports
7. Cassani,C., Vertemara,J., Bassani,M., Marsella,A., Tisi,R., imprecise end joining independently of its catalytic activity. PLoS
Zampella,G. and Longhese,M.P. (2019) The ATP-bound Genet., 9, e1003599.
conformation of the Mre11-Rad50 complex is essential for 30. Lomonaco,S., Bazzano,D. and Wilson,T.E. (2021) Multiple
Tel1/ATM activation. Nucleic Acids Res., 47, 3550–3567. metabolic signals including AMPK and PKA regulate
Downloaded from https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab597/6321018 by guest on 07 December 2021
8. Williams,R.S., Williams,J.S. and Tainer,J.A. (2007) glucose-stimulated double strand break resection in yeast. bioRxiv
Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair doi: https://doi.org/10.1101/2021.02.26.433101, 26 February 2021,
machinery, double-strand break signaling, and the chromatin preprint: not peer reviewed.
template. Biochem. Cell Biol., 85, 509–520. 31. Langmead,B., Trapnell,C., Pop,M. and Salzberg,S.L. (2009) Ultrafast
9. Käshammer,L., Saathoff,J.H., Lammens,K., Gut,F., Bartho,J., and memory-efficient alignment of short DNA sequences to the
Alt,A., Kessler,B. and Hopfner,K.P. (2019) Mechanism of DNA End human genome. Genome Biol., 10, R25.
Sensing and Processing by the Mre11-Rad50 Complex. Mol. Cell, 76, 32. Smith,T., Heger,A. and Sudbery,I. (2017) UMI-tools: modeling
382–394. sequencing errors in unique molecular identifiers to improve
10. Zhang,Y., Hefferin,M.L., Chen,L., Shim,E.Y., Tseng,H.M., Kwon,Y., quantification accuracy. Genome Res., 27, 491–499.
Sung,P., Lee,S.E. and Tomkinson,A.E. (2007) Role of Dnl4-Lif1 in 33. Moreira,J.M., Horz,W. and Holmberg,S. (2002) Neither Reb1p nor
nonhomologous end-joining repair complex assembly and poly(dA*T) elements are responsible for the highly specific chromatin
suppression of homologous recombination. Nat. Struct. Mol. Biol., organization at the ILV1 promoter. J. Biol. Chem., 277, 3202–3209.
14, 639–646. 34. Zhu,Z., Chung,W.H., Shim,E.Y., Lee,S.E. and Ira,G. (2008) Sgs1
11. Wilson,T.E., Grawunder,U. and Lieber,M.R. (1997) Yeast DNA helicase and two nucleases Dna2 and Exo1 resect DNA
ligase IV mediates non-homologous DNA end joining. Nature, 388, double-strand break ends. Cell, 134, 981–994.
495–498. 35. Gobbini,E., Cassani,C., Vertemara,J., Wang,W., Mambretti,F.,
12. Symington,L.S. (2016) Mechanism and regulation of DNA end Casari,E., Sung,P., Tisi,R., Zampella,G. and Longhese,M.P. (2018)
resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol., 51, 195–212. The MRX complex regulates Exo1 resection activity by altering
13. Cannavo,E. and Cejka,P. (2014) Sae2 promotes dsDNA endonuclease DNA end structure. EMBO J., 37. e98588.
activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature, 36. Moreau,S., Ferguson,J.R. and Symington,L.S. (1999) The nuclease
514, 122–125. activity of Mre11 is required for meiosis but not for mating type
14. Chanut,P., Britton,S., Coates,J., Jackson,S.P. and Calsou,P. (2016) switching, end joining, or telomere maintenance. Mol. Cell. Biol., 19,
Coordinated nuclease activities counteract Ku at single-ended DNA 556–566.
double-strand breaks. Nat. Commun., 7, 12889. 37. Cejka,P., Cannavo,E., Polaczek,P., Masuda-Sasa,T., Pokharel,S.,
15. Cejka,P. (2015) DNA end resection: nucleases team up with the right Campbell,J.L. and Kowalczykowski,S.C. (2010) DNA end resection
partners to initiate homologous recombination. J. Biol. Chem., 290, by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and
22931–22938. Mre11-Rad50-Xrs2. Nature, 467, 112–116.
16. Mimitou,E.P. and Symington,L.S. (2008) Sae2, Exo1 and Sgs1 38. Gobbini,E., Casari,E., Colombo,C.V., Bonetti,D. and Longhese,M.P.
collaborate in DNA double-strand break processing. Nature, 455, (2020) The 9-1-1 complex controls Mre11 nuclease and checkpoint
770–774. activation during short-range resection of DNA double-strand
17. Garcia,V., Phelps,S.E., Gray,S. and Neale,M.J. (2011) Bidirectional breaks. Cell Rep., 33, 108287.
resection of DNA double-strand breaks by Mre11 and Exo1. Nature, 39. Marini,F., Rawal,C.C., Liberi,G. and Pellicioli,A. (2019) Regulation
479, 241–244. of DNA double strand breaks processing: focus on barriers. Front.
18. Bonilla,B., Hengel,S.R., Grundy,M.K. and Bernstein,K.A. (2020) Mol. Biosci., 6, 55.
RAD51 gene family structure and function. Annu. Rev. Genet., 54, 40. Sorenson,K.S., Mahaney,B.L., Lees-Miller,S.P. and Cobb,J.A. (2017)
25–46. The non-homologous end-joining factor Nej1 inhibits resection
19. Reginato,G., Cannavo,E. and Cejka,P. (2017) Physiological protein mediated by Dna2-Sgs1 nuclease-helicase at DNA double strand
blocks direct the Mre11-Rad50-Xrs2 and Sae2 nuclease complex to breaks. J. Biol. Chem., 292, 14576–14586.
initiate DNA end resection. Genes Dev., 31, 2325–2330. 41. Mojumdar,A., Sorenson,K., Hohl,M., Toulouze,M.,
20. Wang,W., Daley,J.M., Kwon,Y., Krasner,D.S. and Sung,P. (2017) Lees-Miller,S.P., Dubrana,K., Petrini,J.H.J. and Cobb,J.A. (2019)
Plasticity of the Mre11-Rad50-Xrs2-Sae2 nuclease ensemble in the Nej1 interacts with Mre11 to regulate tethering and Dna2 binding at
processing of DNA-bound obstacles. Genes Dev., 31, 2331–2336. DNA double-strand breaks. Cell Rep., 28, 1564–1573.
21. Cannavo,E., Reginato,G. and Cejka,P. (2019) Stepwise 5’ DNA 42. Gobbini,E., Vertemara,J. and Longhese,M.P. (2018) Local unwinding
end-specific resection of DNA breaks by the Mre11-Rad50-Xrs2 and of double-strand DNA ends by the MRX complex promotes Exo1
Sae2 nuclease ensemble. PNAS, 116, 5505–5513. processing activity. Mol. Cell. Oncol., 5, e1511208.
22. Peng,H., Zhang,S. and Chen,X. (2021) Monitoring 5’-end resection 43. Yu,T.Y., Garcia,V.E. and Symington,L.S. (2019) CDK and
at site-specific double-strand breaks by Southern blot analysis. Mec1/Tel1-catalyzed phosphorylation of Sae2 regulate different
Methods Mol. Biol., 2196, 245–255. responses to DNA damage. Nucleic Acids Res., 47, 11238–11249.
23. Zhou,Y., Caron,P., Legube,G. and Paull,T.T. (2014) Quantitation of 44. Wang,W., Daley,J.M., Kwon,Y., Xue,X., Krasner,D.S., Miller,A.S.,
DNA double-strand break resection intermediates in human cells. Nguyen,K.A., Williamson,E.A., Shim,E.Y., Lee,S.E. et al. (2018) A
Nucleic Acids Res., 42, e19. DNA nick at Ku-blocked double-strand break ends serves as an entry
24. Mimitou,E.P., Yamada,S. and Keeney,S. (2017) A global view of site for exonuclease 1 (Exo1) or Sgs1-Dna2 in long-range DNA end
meiotic double-strand break end resection. Science, 355, 40–45. resection. J. Biol. Chem., 293, 17061–17069.
25. Mimitou,E.P. and Keeney,S. (2018) S1-seq assay for mapping 45. Yu,T.Y., Kimble,M.T. and Symington,L.S. (2018) Sae2 antagonizes
processed DNA ends. Methods Enzymol., 601, 309–330. Rad9 accumulation at DNA double-strand breaks to attenuate
26. Brachmann,C.B., Davies,A., Cost,G.J., Caputo,E., Li,J., Hieter,P. and checkpoint signaling and facilitate end resection. PNAS, 115,
Boeke,J.D. (1998) Designer deletion strains derived from E11961–E11969.
Saccharomyces cerevisiae S288C: a useful set of strains and plasmids 46. Li,K., Bronk,G., Kondev,J. and Haber,J.E. (2020) Yeast ATM and
for PCR-mediated gene disruption and other applications. Yeast, 14, ATR kinases use different mechanisms to spread histone H2A
115–132. phosphorylation around a DNA double-strand break. PNAS, 117,
27. Wu,D., Topper,L.M. and Wilson,T.E. (2008) Recruitment and 21354–21363.
dissociation of nonhomologous end joining proteins at a DNA 47. Rahal,E.A., Henricksen,L.A., Li,Y., Williams,R.S., Tainer,J.A. and
Dixon,K. (2010) ATM regulates Mre11-dependent DNAYou can also read

















































