MPhil/Part III Neural algorithmic reasoning with warm starts
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
[MPhil/Part III] Neural algorithmic reasoning with
warm starts
Prof Pietro Lio (pl219) and Dobrik Georgiev, dgg30@cam.ac.uk
Neural algorithmic reasoning (NAR) is the art of building neural networks that are able to
execute algorithmic computation (https://arxiv.org/abs/2105.02761). Some of these method’s
benefits are end-to-end differentiability and/or being able to deploy ‘deterministic’ algorithms
to ‘new’ scenarios without the need for hand-writing the abstraction of reality. (An example is
https://arxiv.org/abs/2010.13146) However NAR is limited to ‘mimicking’ the algorithm and is
bound by the original algorithm’s complexity (e.g. you cannot find shortest paths in less time
compared to a classical algorithm).
Another line of research investigates reducing the worst-case complexity bounds of
algorithms. A couple of recent papers https://arxiv.org/pdf/2204.12055.pdf (ICML 2022),
https://arxiv.org/pdf/2107.09770.pdf use “machine learning” to predict dual variables of a
linear programming formulation of the algorithms. The predicted dual solution is then
rounded to an optimal dual solution in a lower time-complexity compared to the original
algorithm.
A potential project can investigate whether the two approaches can work synergistically. For
example, a prediction on the dual linear programming formulation can be converted to a
primal solution, which can be used to instantiate the input to the NAR network.
Further extensions are also possible:
● Currently the last two LP papers make their prediction by taking the average of the
optimal dual variables in the training sets. Although, according to the theory in the
papers, this results in lowest L1 loss in expectation, in practice using a NN predictor
(e.g. a shallow MPNN or a cheap GNN like https://arxiv.org/pdf/1902.07153.pdf ) may
result in better predictions that take into account topology.
● NAR is more robust to noise in the data due to representing information in vector
space. Instead of predicting actual scalars, we can try directly feeding in an
embedding of the scalar, e.g. if we decode the scalar from a vector representation
computed from a neural network, we can instead feed the vectorial representation.
[MPhil/Part III] Neural Algorithms for Subgraph
Neural Networks
Prof Pietro Lio (pl219) and Dobrik Georgiev, dgg30@cam.ac.uk
Deep learning on graphs (a.k.a. GNNs) has received a lot of attention in the past few years.
However, despite this attention, GNNs mainly focus on learning good node/edge/graph
representations for solving node/edge/graph tasks, leaving aside tasks on subgraphs. One
(relatively) new line of work introduces SubGNN, a framework which identifies six key
properties of subgraphs and proposes a specifically designed message passing mechanism
for encoding these properties.
One part of this mechanism is a similarity function between a subgraph component and an
anchor patch (have a glimpse at the paper for the terminology, esp. Figure 1 and chapters
3;4). For some channels, this similarity function is estimated as the inverse average shortest
path length, for others it’s the normalised dynamic time warping (a Dynamic Programming
algorithm). These similarity functions are not learnable and return a scalar value. It might be
interesting to experiment with making a learnable, yet faithful to the encoding original
properties \gamma. (N.B. The paper itself proposes a learnable \gamma as a model
extension).
One way this could be achieved is Neural Algorithmic Reasoning. Neural algorithmic
reasoning (NAR) is the art of building neural networks that are able to execute algorithmic
computation (https://arxiv.org/abs/2105.02761). Some of these method’s benefits are
end-to-end differentiability and/or being able to deploy ‘deterministic’ algorithms to ‘new’
scenarios without the need for hand-writing the abstraction of reality. (An example is
https://arxiv.org/abs/2010.13146). A potential project would investigate:
● Can we learn shortest path \gamma with a NAR-oriented GNN? This would require
learning to perform Floyd-Warshal (dataset available through CLRS-30 or could be
synthesised manually).
● Can we use this learnt \gamma when training/using SubGNN? A transfer learning
approach similar to https://arxiv.org/abs/2110.14056 could be used here.
● Instead of using a scalar when multiplying the computed message, a channel-wise
product can with a vector can be used. This would be the vector from which \gamma
is decoded. (If necessary the vector could be forced to have, e.g. only positive
values)
● Can we perform the above three on the DWT \gamma.
Training would be performed with manually synthesised datasets (as and where needed),
datasets from CLRS-30 and datasets from SubGNN. Evaluation would be performed on the
datasets provided by SubGNN. Our baselines would be ‘ground-truth’ algorithms (i.e.
comparing how much we deviate from the g.t. \gamma), as well as SubGNN itself.[MPhil/Part III] Learning Symmetries / Breaking
Symmetries in Geometric Deep Learning
Prof Pietro Lio (pl219), Chaitanya K. Joshi (ckj24@cam.ac.uk),
Simon Mathis, Alex Norcliffe, Charlie Harris
Geometric Deep Learning provides a unified framework for thinking about a broad class of
deep learning architectures from the perspectives of symmetry and invariance. E.g.
Convolutional Networks for computer vision are translation invariant, while Graph Neural
Networks and Transformers for structured datasets are permutation invariant. However,
designing these architectures assumes the existence of exact and global symmetries acting
on our data.
In this project, we will empirically and/or theoretically question this idea: do we need exact
symmetries and equivariance, or can we build more expressive and performant deep
learning models without ‘baking in’ exact symmetries?
Interesting papers and articles:
- Learning Invariances in Neural Networks: https://arxiv.org/abs/2010.11882
- Approximately Equivariant Networks for Imperfectly Symmetric Dynamics:
https://arxiv.org/abs/2201.11969
- Naturally Learnt Equivariance in CNNs: https://distill.pub/2020/circuits/equivariance/[MPhil/Part III] Interpretability of Graph Neural Networks from the lens of Circuits Prof Pietro Lio (pl219), Chaitanya K. Joshi (ckj24@cam.ac.uk), Pietro Barbiero, Charlotte Magister Circuits are a neural network interpretability technique introduced by OpenAI for ConvNets in 2020. The team has since started its own company based on this work - Anthropic AI. They recently received funding worth US$ ~600 Million, so they must be on to something very interesting? Anthropic has recently been working on Circuits for Transformers, which are a type of neural network which processes sets or tokens (such as words in a sentence) by building all pairwise relationships among them. Transformers have become the go-to architecture for natural language processing, and may emerge as a modality-agnostic, one-size-fits-all neural network architecture. Interestingly, there are deep connections between Transformers and Graph Neural Networks. This project will analyse GNNs through the lens of the Circuits framework, similar to how Anthropic is analysing Transformers. The harmony between Transformers and GNNs may lead our findings to be generally relevant for advancing interpretability and understanding of both classes or architectures (or are they the same class of architectures ;)).
[MPhil/Part III] Expressive Power of Graph Neural Networks for Graph Generation Prof Pietro Lio (pl219) and Chaitanya K. Joshi (ckj24@cam.ac.uk) The theoretical expressive power of Graph Neural Networks (GNNs) has been studied extensively in recent years, especially through the lens of graph isomorphism testing [1]. This line of work has lead to significant advances in practical GNN modelling [2, 3]. However, all these works have focused on predictive models, i.e. given an input graph X, output some property Y. In this project, we will study the impact of GNN expressivity on the inverse process of graph generation and graph inverse design, i.e. given some desired property Y, generate a graph X that satisfies this property. We will study this problem from an empirical and/or theoretical angle. Possible outcomes of this project would be: provably powerful graph generative models, deeper understanding of GNN expressivity on generative modelling, hands-on experience with latest GNNs and graph generation tools. Why graph generation? Graph generation is interesting beyond typical GNN tasks (node/link prediction) as it requires building an entire graph with multiple nodes and edges. Advances in generative modelling of graphs can have significant positive impact in AI for scientific discovery – de-novo inverse design is the ‘holy grail’ of AI for science.
[MPhil/Part III] Theory of Geometric Graph Neural Networks Prof Pietro Lio (pl219), Chaitanya K. Joshi (ckj24@cam.ac.uk), Simon Mathis In an upcoming paper, we have studied the expressive power of a new and emerging class of GNN architectures specialised for 3D geometric objects: https://openreview.net/pdf?id=Rkxj1GXn9_ In this project, we will build upon these theoretical foundations and study geometric GNNs in more detail. We may focus our attention on the concept of Geometric Computation Trees, and follow the methodology of this seminal paper from Garg-etal: http://proceedings.mlr.press/v119/garg20c.html. Key outcomes would include deeper understanding of what this new class of GNNs can and cannot do, as well as their generalisation to new data. As this would be theoretical work, it would involve less coding and more pen-and-paper work, with some synthetic experimentation that can be run to supplement the theory.
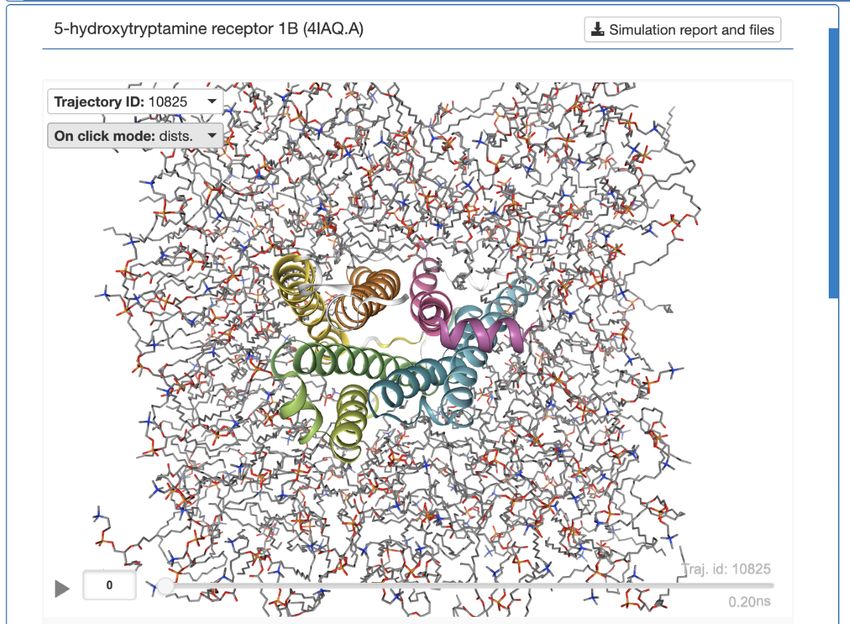
[MPhil/Part III] Learning to Simulate GPCR Protein
Dynamics
Prof Pietro Lio (pl219), Chaitanya K. Joshi (ckj24@cam.ac.uk),
Charles Harris
Following the recent success of using Geometric Deep Learning and Graph Neural Networks
for simulating the dynamics of small molecules, it is natural to ask whether we can translate
this success to macromolecules such as proteins.
In this project, we will focus on a class of proteins called G-Protein Coupled Receptors
(GPCRs) which are important drug targets for cancer. We will work with a community
repository of GPCR dynamics: https://submission.gpcrmd.org/home/ and the primary
milestone of the project will be to make this data ‘machine learning-ready’.
We -canHow
thenwell
ask can GNNs
several learn to simulate
interesting protein
questions, dynamics?
such as:
- Can we use protein dynamics trajectories to improve the prediction of ligand binding
affinity prediction for GPCRs?
A sample datapoint: the dataset provides 500ns trajectories of several GPCRs as well as the
solvent/membrane around these proteins (you can toggle it via the tab 'Structure selection').[MPhil/Part III] CRISPR Interference: Guide Design Prof Pietro Lio (pl219) and Jacob Moss (jm2311@cam.ac.uk) CRISPR gene editing is a breakthrough in genetic engineering, with uses such as gene therapy and high-throughput perturbation analysis. CRISPR interference (CRISPRi) [1] is a relatively new technique for gene interaction mapping. This project looks at optimal experimental design via the probabilistic ranking of CRISPRi guides and analysis of individual guide efficacy. It explores the systems biology context of chromatin features, proximity of guide to target promoter and TSS, as well as underlying transcriptional activity in that cell line. There are several publicly available pooled CRISPRi datasets, namely [2], which can be used for understanding the expected outcome for a given guide pair. Given this expectation, the aim would be to identify factors impeding a guide’s performance. References: [1] Lim et al., https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3664290/ [2] Horlbeck et al., https://www.cell.com/cell/pdf/S0092-8674(18)30735-9.pdf
[MPhil/Part III] Advancing neural
aggregation/readout functions in GNNs
Prof Pietro Lio (pl219) and David Buterez (db804@cam.ac.uk)
In one of our recent papers (to appear soon), we introduced neural aggregation (readout)
functions for GNNs, in the form of various kinds of neural networks that replace the static
and simple functions that are most commonly used in GNNs, such as sum, mean, or
maximum. So far, we have mostly focused on performance gains and trade-offs regarding a
lack of permutation invariance in the function.
As neural readouts introduce a new neural network component to GNNs, this creates
opportunities to extend the existing methodology and study their behaviour in different
conditions. Two possible starting points for further study are:
1. Hybrid readouts – Instead of choosing a single readout function type for a given
GNN, we can instead use multiple functions at the same time, and combine the
outputs in a simple way (e.g. concatenation). This would be interesting from two
angles: (a) see if there is any performance gain, for example by making it easier for
the GNN to learn, and (b) deduce if the properties learnt by the different readouts are
similar (i.e. they learn the same things) or they can potentially learn different patterns.
In the latter case, they might cover each other’s weakness, working synergistically,
and lead to more robust and stronger models. Another aspect to look into is
weighting the different readouts, which might be beneficial to performance/learning.
2. Pretraining readouts – We observed some of the largest benefits on molecular
regression datasets (e.g. QM9). Thus, one question that we can ask is if pretraining
on a set of simple chemical properties (easily computable with RDKit for example) is
beneficial to the main prediction task, for example the QM9 properties. Pre-training
for GNNs was discussed in the literature [2], however not in the context of neural
readouts, where the observed behaviours could be completely different.
Another strategy could be to pretrain the GNN on similar but cheaper-to-compute
molecular measurements, for example quantum properties measured at a less
expensive level of theory, followed by training on the higher-quality labels. This can
be done on a dataset such as QMugs [1].
For the project itself, we will focus on formulating valid scientific questions based on the
narrative described above and answering them with appropriate experiments. A good initial
dataset to focus on initially is QM9. Other potential ideas to incorporate in this project are:
1. Studying what the attention in the attention-based neural readout learns, and if this
can be related to the chemical properties in any way.
2. Studying how the neural readouts alter or change the molecular embeddings when
using chemical-reaction aware models [3]Depending on the amount of work that we cover (including any time after the thesis is submitted) and the results, we can think about publishing a paper to either a more chemically-oriented venue or a more general ML venue. [1] QMugs, quantum mechanical properties of drug-like molecules [2] Does GNN Pretraining Help Molecular Representation? [3] Chemical-Reaction-Aware Molecule Representation Learning
[MPhil/Part III] Residue identity prediction from an
amino acid residue’s atomic-environment with
equivariant graph machine learning
Prof Pietro Lio (pl219) , Simon Mathis (svm34@cam.ac.uk),
Chaitanya Joshi, Charlie Harris
Context:
Protein engineering is the process of mutating a protein’s amino-acid sequence in certain
targeted positions in order to optimise a set of functional characteristics. For instance, a
common target is to increase a protein’s thermal stability. Recently, machine learning is
increasingly used to identify likely beneficial points in the protein sequence to mutate. To
give just one example, a 3D-CNN based approach [1] was recently used to engineer plastic
decomposing enzymes for higher thermal stability [2].
While 3D-CNNs hold the state-of-the art for predicting the most promising amino-acid given
the local atomic environment, they require voxelising the atomic environment and working
with cubic domains [1,2]. Recent advances in equivariant graph neural network [3] modelling
show promise to overcome these problems and could lead to way to more accurate
identification of the best fitting amino acid residues for a given atomic environment.
This project aims to investigate whether equivariant graph neural networks can outperform
3D-CNNs (the current state-of-the-art) in the residue identity prediction and improve protein
engineering practice.
- Given the atomic environment of a position in the protein, use an equivariant graph
Task:
neural network model to predict the most likely amino acid residue (among the 20
natural amino acids) that would fit this environment.
Data:
- Initial data to get started is available from the atom3d RES benchmark dataset [4].
This data includes about 3 Mio. datapoints from over 24’000 protein structures from
the Protein Data Bank.
Each data point is a collection of atom coordinates (x,y,z) and atom type (C, N, O, S)
in a 20 Angstrom ball around the position of the amino acid to predict. The prediction
target is a label of one of the 20 amino acids [multi-class classification task].
Further reading:
- [1] Torng & Altman 2017 - 3D deep convolutional neural networks for amino acid
environment similarity analysis [pubmed]
- [2] Lu et al. 2022 - Machine learning-aided engineering of hydrolases for PET
depolymerization [nature]
- [3] Schütt et al. 2021 - PaiNN: Equivariant message passing for the prediction of
tensorial properties and molecular spectra [arxiv]
- [4] Townshend et al. 2021 - Atom3D: Tasks On Molecules in Three Dimensions
[arxiv][MPhil/Part III] Multi-body equivariant graph
modelling for biochemical reaction parameter
prediction
Prof Pietro Lio (pl219) , Simon Mathis (svm34@cam.ac.uk), Charlie
Harris (cch57@cam.ac.uk)
Context: Predicting biochemical reaction parameters such as the substrate affinities K_m or
reaction turnover rates k_cat are inherent multi-body problems. They depend on the
interaction between multiple molecules: a protein and one or more substrates and products.
Current predictive models for reaction parameters [1, 2] do not take the geometry of the
constituent molecules into account. The recent advances in equivariant graph machine
learning [3] and the release of 200 Mio. predicted protein structures in the AlphaFold
database [4] indicate the potential to bring reaction parameter prediction to the next level by
utilising 3D molecular information.
This project aims to assess whether the recently available, predicted protein structures from
AlphaFold2 can be used in conjunction with 3D models of small molecules to improve the
task of reaction parameter prediction.
Task:
Given the (predicted) structure of a protein and its substrates and products, predict the
substrate affinity (K_m) and turnover rate (k_cat). [regression problem]
(Extension: Use binding problem data and investigate transfer to catalysis problem)
Data:
- An enzyme kinetics dataset with 10’000+ entries of reaction parameters and
reactants (proteins & small molecules) was curated and pre-processed in-house and
is available for the student. Protein structures and molecular descriptors (SMILES)
are already contained in the dataset.
Further reading:
- [1] Kroll et al. 2021 - Deep learning allows genome-scale prediction of Michaelis
constants from structural features.
[https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3001402]
- [2] Li et al. 2021 - Deep learning based kcat prediction enables improved enzyme
constrained model reconstruction [https://doi.org/10.1101/2021.08.06.455417]
- [3] Stärk et al. 2022 - EquiBind [https://arxiv.org/pdf/2202.05146.pdf]
- [4] https://alphafold.ebi.ac.uk
- [5] Hunter, 1993 - Molecular biology for computer scientists
[https://www.aaai.org/Papers/Hunter/01-Hunter.pdf] (especially chapter 4)[MPhil/Part III] Self-supervised learning of
protein-ligand interactions from unlabeled data
Prof Pietro Lio (pl219) , Charlie Harris (cch57@cam.ac.uk), Simon
Mathis
Context: Understanding the interactions between protein and ligand molecules is a
fundamental problem in biology and small molecule drug discovery [1]. Many approaches try
to model protein-ligand interactions using machine learning but all struggle with the lack of
3D data samples of a ligand molecule bound into a protein receptor. Furthermore, the data
we do have only covers a very small fraction of chemical space, meaning models do not
generalise well to new classes of molecules. A recent trend in other ML fields is to pretrain
large models on massive amounts of unlabeled data. These models then serve as generalist
‘foundation models’ [2] that, starting with a broad understanding of the problem domain, can
then be fine tuned for specific tasks using a small amount of labelled data. Whilst being very
successful in language learning and protein sequences [3], the same approaches have only
been applied modestly to protein structure [4].
Task: The student will design and train a model that will be pretrained to understand protein-
ligand interactions using self-supervised learning with the eventual goal being that this
foundation model can then be fine-tuned for specific tasks (e.g. ligand binding affinity
prediction). The small amount of 3D structural data available will be used to validate
unsupervised learning of protein-ligand interactions (e.g. by attention weight analysis).
Data: The student will primarily use datasets relating to protein-ligand interactions. This will
fall into (i) structurally-unlabelled protein-ligand interaction data, large datasets of
2 classes:
protein-ligand pairs for which we do not know the 3D structure of the binding
interactions (e.g. BindingDB) and
(ii) structurally-labelled datasets of protein-ligand interactions where we know the 3D
binding interaction (e.g. PDBBind).
Further reading:
- [1] Insights into protein–ligand interactions: mechanisms, models, and methods
[https://link.springer.com/content/pdf/10.1007/978-1-62703-398-5.pdf]
- [1] On the opportunities and risks of Foundations Models
[https://arxiv.org/abs/2108.07258]
- [2] Biological structure and function emerge from scaling unsupervised learning to
250 million protein sequences [https://www.biorxiv.org/content/10.1101/622803v4]
- [3] Protein Representation Learning by Geometric Structure Pretraining
[https://arxiv.org/abs/2203.06125][MPhil/Part III] Geometric deep learning for protein
normal mode dynamics
Prof Pietro Lio (pl219) , Simon Mathis (svm34@cam.ac.uk), Charlie
Harris, Arian Jamasb
Context:
Protein dynamics are key to understanding and characterising protein function [1]. While
there has been much attention on deep learning for static protein structures, little work has
been done to incorporate information on protein dynamics into protein representations. This
work focuses on exploring the use of protein normal modes, a well-known classical technique
to interrogate (protein) vibrational modes, in modern deep learning workflows. Recent work
on incorporating eigenvalues and eigenspaces into equivariant geometric graph
representations [2,3] is a promising way to combine structural and normal mode data and
provides a principled way to improve on previous attempts to leverage protein normal modes
[4]. In this project, we will assess to which extent including normal-mode data can improve
protein function prediction and protein domain motion classification.
- Experimental protein structural data (100k+ structures) is available from the protein
Data:
data bank (PDB) and can be used with pre-processing scripts available from the
supervisors to extract protein normal modes.
- Protein functional classification data (EC / GO annotation) is available for all PDB
structures.
- Data from the Protein structural change database (PSCDB) will be available for the
protein domain motion classification task.
Further reading:
- [1] Hunter, 1993 - Molecular biology for computer scientists
[https://www.aaai.org/Papers/Hunter/01-Hunter.pdf] (especially chapter 4)
- [2] Lim et al. 2022 - Sign and basis invariant networks for spectral graph
representation learning [https://arxiv.org/abs/2202.13013]
- [3] Satorras et al. 2021 - E(n) equivariant graph neural networks
[https://arxiv.org/pdf/2102.09844.pdf]
- [4] Chiang et al. 2022 - Encoding protein dynamic information in graph representation
for functional residue identification [https://arxiv.org/pdf/2112.12033.pdf][MPhil/Part III] Scalable Logic Explained Networks for Concept Embeddings Prof Pietro Lio (pl219) and, Pietro Barbiero (pb737@cam.ac.uk), Francesco Giannini, Gabriele Ciravegna, Mateo Espinoza Zarlenga Context Deploying AI-powered systems requires trustworthy models supporting effective human interactions, going beyond raw prediction accuracy. Concept bottleneck models [1] promote trustworthiness by conditioning classification tasks on an intermediate level of human-like concepts [2]. This enables human interventions which can correct mispredicted concepts to improve the model's performance [1]. However, existing concept bottleneck models are unable to find optimal compromises between high task accuracy, robust concept-based explanations, and effective interventions on concepts---particularly in real-world conditions where complete and accurate concept supervisions are scarce. The recent NeurIPS paper “Concept Embedding Models” [3] addresses this trade-off by learning interpretable high-dimensional concept representations. However, finding global logic explanations (as in Logic Explained Networks [4]) for Concept Embedding Models is still an open challenge. Moreover, the extraction of logic explanations does not scale well for large data sets. In particular the aggregation of minterms grows rapidly with the number of samples. Research question Design a scalable Logic Explained Network [4] for concept embeddings [3]. Possible research direction Design a layer within the Logic Explained Network to cluster similar samples for a faster minterm aggregation (e.g., following [5]). [1] Koh, Pang Wei, et al. "Concept bottleneck models." International Conference on Machine Learning. PMLR, 2020. [2] Kim, B., Wattenberg, M., Gilmer, J., Cai, C., Wexler, J., & Viegas, F. (2018, July). Interpretability beyond feature attribution: Quantitative testing with concept activation vectors (tcav). In International conference on machine learning (pp. 2668-2677). PMLR. [3] Zarlenga, M. E., Barbiero, P., Ciravegna, G., Marra, G., Giannini, F., Diligenti, M., ... & Jamnik, M. (2022). Concept Embedding Models. arXiv preprint arXiv:2209.09056. [4] Barbiero, P., Ciravegna, G., Giannini, F., Lió, P., Gori, M., & Melacci, S. (2022, June). Entropy-based logic explanations of neural networks. In Proceedings of the AAAI Conference on Artificial Intelligence (Vol. 36, No. 6, pp. 6046-6054). [5] Magister, L. C., Barbiero, P., Kazhdan, D., Siciliano, F., Ciravegna, G., Silvestri, F., ... & Lio, P. (2022). Encoding Concepts in Graph Neural Networks. arXiv preprint arXiv:2207.13586.
[MPhil/Part III] Self-Explainable Graph Neural Networks via Concept Lattices Prof Pietro Lio (pl219) and, Pietro Barbiero (pb737@cam.ac.uk), Lucie Charlotte Magister, Francesco Giannini Context The opaque reasoning of Graph Neural Networks (GNNs) induces a lack of human trust. Existing graph network explainers attempt to address this issue by providing post-hoc explanations, however, they fail to make the model itself more interpretable. The “Concept Encoder Module” [1] aims to address this problem by allowing GNNs to discover concepts [2] (i.e., high-level units of information represented as subgraph motifs) and use them to solve the task during training. However, the Concept Graph Module can only generate a single concept for each input graph, while in general a graph can be composed of multiple concepts organised in hierarchies (e.g., lattices) [3]. Moreover, like most GNNs explanation methods, this module was designed for graph and node classification, while GNNs explanations for tasks such as link prediction are mostly unexplored. Research question Design an unsupervised concept encoder for GNNs generating a concept lattice to solve classical GNN tasks. Possible research direction Use the Concept Encoder Module to generate a concept lattice using Formal Concept Analysis [3]. [1] Magister, L. C., Barbiero, P., Kazhdan, D., Siciliano, F., Ciravegna, G., Silvestri, F., ... & Lio, P. (2022). Encoding Concepts in Graph Neural Networks. arXiv preprint arXiv:2207.13586. [2] Kim, B., Wattenberg, M., Gilmer, J., Cai, C., Wexler, J., & Viegas, F. (2018, July). Interpretability beyond feature attribution: Quantitative testing with concept activation vectors (tcav). In International conference on machine learning (pp. 2668-2677). PMLR. [3] Ganter, B., & Wille, R. (2012). Formal concept analysis: mathematical foundations. Springer Science & Business Media.
You can also read

















































