Integrative Approaches to Enhance Understanding of Plant Metabolic Pathway Structure and Regulation1
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Update on Integrative Studies
Integrative Approaches to Enhance Understanding of
Plant Metabolic Pathway Structure and Regulation1
Takayuki Tohge*, Federico Scossa, and Alisdair R. Fernie
Max-Planck-Institute of Molecular Plant Physiology, 14476 Potsdam-Golm, Germany (T.T., A.R.F.); and
Consiglio per la Ricerca e Analisi dell’Economia Agraria, Centro di Ricerca per la Frutticoltura, 00134 Rome,
Italy (F.S.)
Downloaded from https://academic.oup.com/plphys/article/169/3/1499/6113889 by guest on 28 January 2022
Huge insight into molecular mechanisms and biological network coordination have been achieved following the application of
various profiling technologies. Our knowledge of how the different molecular entities of the cell interact with one another
suggests that, nevertheless, integration of data from different techniques could drive a more comprehensive understanding of
the data emanating from different techniques. Here, we provide an overview of how such data integration is being used to aid
the understanding of metabolic pathway structure and regulation. We choose to focus on the pairwise integration of large-scale
metabolite data with that of the transcriptomic, proteomics, whole-genome sequence, growth- and yield-associated phenotypes,
and archival functional genomic data sets. In doing so, we attempt to provide an update on approaches that integrate data
obtained at different levels to reach a better understanding of either single gene function or metabolic pathway structure and
regulation within the context of a broader biological process.
The diversity of metabolites in the plant kingdom is utility in enhancing our understanding of enzyme
staggering: a commonly quoted estimate is that plants mechanisms (and their regulation) and about the in
produce somewhere in the order of 200,000 unique vivo functions of enzymes, respectively. To give just a
chemical structures (Dixon and Strack, 2003; Yonekura- couple of recent examples from organic acid metabo-
Sakakibara and Saito, 2009; Tohge et al., 2014). Of these, lism, a detailed study of the effect of phosphorylation of
only a relatively small subset will be abundant in any phosphoenolpyruvate carboxylase reveals an important
given tissue or any one species (Fernie, 2007); however, anaplerotic control point in developing castor bean
certain species have evolved a particularly rich meta- (Ricinus communis) endosperm (Hill et al., 2014), while
bolic diversity, presumably in response to environ- the enzyme pyruvate orthophosphate dikinase was
mental features of their habitat (for examples, see recently demonstrated to represent a second gateway
Futuyma and Agrawal, 2009; Moore et al., 2014; Li et al., for organic acids into the gluconeogenic pathway in
2015). Given these facts, it is unsurprising that our Arabidopsis (Arabidopsis thaliana; Eastmond et al., 2015).
current understanding of the metabolic structure of a We aim to provide examples from both primary and
large number of pathways remains fragmentary; not to secondary metabolism and to illustrate the power of
mention our current views of regulatory mechanisms such approaches both in (1) gene functional annotation
underlying metabolite accumulation, which cover, at and (2) enhancing our understanding of the systems-
best, a very limited fraction of the metabolic network. level response to cellular circumstances. We will addi-
This statement is especially true for the highly special- tionally discuss recent studies combining genome
ized pathways of secondary metabolism, although a sequence data with metabolomics in order to highlight
number of gaps still remain to be filled also concerning the utility of such approaches in metabolic quantitative
important sectors of plant primary metabolism. As loci analyses. Finally, we will detail insight that can be
detailed in other Update articles within this issue, the obtained from fusing archived data that can be down-
adoption of various broad-scale profiling technolo- loaded from databases with experimental data gener-
gies to assess the gene, transcript, protein, and small ated de novo. Given that, as documented previously
molecule complement of the cell has started to mine (Fernie and Stitt, 2012), a number of complicating fac-
this metabolic complexity. Additionally, the same ap- tors still exist when attempting such analyses, we will
proaches have also started to shed light on the evolu- discuss these on an approach-by-approach basis.
tion of gene and metabolite regulatory networks across
the plant kingdom. In addition to large-scale profiling
approaches, classical reductionist biochemistry and
reverse genetic approaches retain, in any case, great INTEGRATING METABOLITE AND
TRANSCRIPTOME DATA
1
This work was supported by the Max Planck Society (to T.T. and The earliest integrative approaches with relevance to
A.R.F.) and an Alexander von Humboldt grant (to T.T.). plant metabolism featured the combination of data
* Address correspondence to tohge@mpimp-golm.mpg.de. from transcript and metabolite profiling (Urbanczyk-
www.plantphysiol.org/cgi/doi/10.1104/pp.15.01006 Wochniak et al., 2003; Achnine et al., 2005; Tohge
Plant PhysiologyÒ, November 2015, Vol. 169, pp. 1499–1511, www.plantphysiol.org Ó 2015 American Society of Plant Biologists. All Rights Reserved. 1499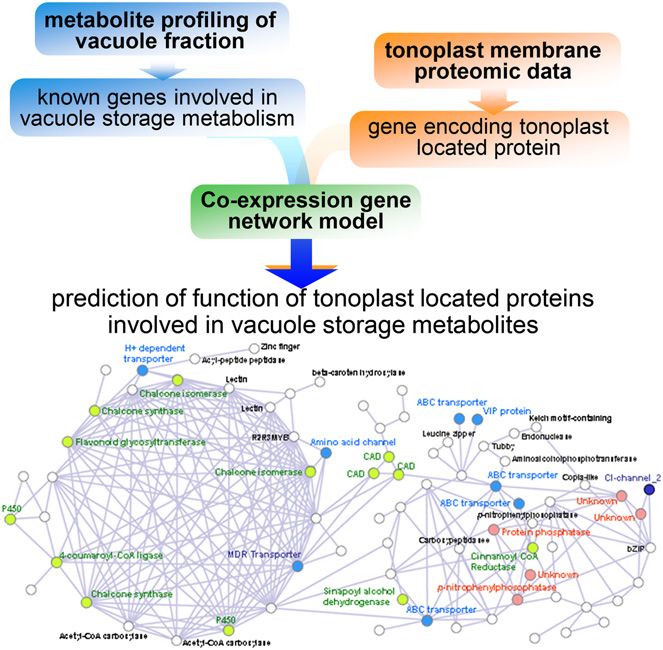
Tohge et al.
et al., 2005). Such studies were initially restricted to metabolites across tuber development, irrespective of
model species for which ESTs or oligonucleotides were whether the transcript was associated with the meta-
available; early transcriptomics approaches relied in bolic pathway under question or not (Urbanczyk-
fact on differential hybridization of complementary Wochniak et al., 2003).
DNA samples to known sequences immobilized on This approach was indeed able to identify some
solid supports. The advent of next-generation se- transcripts that exhibited very high correlation with the
quencing technologies, however, has removed this expression of certain genes and, as such, proved effec-
barrier, and far more exotic species are beginning to be tive in identifying a number of candidate genes for
studied using this approach (Góngora-Castillo et al., biofortification. By corollary, the same approach can be,
2012; Gechev et al., 2013; Li et al., 2015). Two basic and indeed has been, used to elucidate the variation in
questions are commonly addressed by combining tran- gene-to-metabolite networks following short- and long-
script and metabolite data. The first concerns whether a term nutritional stresses in Arabidopsis (Hirai et al.,
Downloaded from https://academic.oup.com/plphys/article/169/3/1499/6113889 by guest on 28 January 2022
gene functions within a given metabolic pathway. 2004) or to identify metabolic regulators of gene ex-
When a better characterization of the pathway is achieved, pression (Hirai et al., 2007). Cryptoxanthin, for exam-
it becomes fundamental to investigate also the extent ple, was identified as highly correlating with a broad
of transcriptional control (except in some cases, for ex- number of genes across diverse environmental condi-
ample, regulation by posttranscriptional modifications tions in Arabidopsis (Hannah et al., 2010), and the or-
of the enzyme and positive/negative feedback reg- ganic acid malate was putatively identified (Carrari
ulation by substrates/products) under various physio- et al., 2006) and subsequently confirmed (Centeno et al.,
logical conditions and how it is distributed across the 2011) to be important in mediating the ripening process
various enzymatic steps. in tomato (Solanum lycopersicum). Such current studies
Initial observations about the role of differential gene are all examples of the guilt-by-association approach,
expression in tuning the synthesis of metabolites date which in essence postulates biological entities as being
back to the 1990s. Some specific pathways, such as functionally related if they exhibit strong correlation or
hormone, glucosinolate, and flavonoid biosynthesis, coresponse across a wide range of cellular circum-
were the initial focus of these investigations. For ex- stances. The power of this approach is that, given that it
ample, differential mechanisms of gene expression does not rely on a priori pathway knowledge, it can
helped clarify in Arabidopsis the involvement of two have great utility in identifying novel metabolic inte-
different nitrilase genes in regulating the synthesis of gration and/or novel regulatory mechanisms (Hirai
auxin (Bartling et al., 1994). Similarly, the contributions et al., 2007; Tohge et al., 2007; Yonekura-Sakakibara
of gene duplication and inducible gene expression et al., 2008; Tohge and Fernie, 2010). However, a
(differential activation of subsets of biosynthetic genes) drawback of the approach is that, in the absence of
were shown to impact the amount and the composition subsequent rounds of experimentation, it is difficult to
of glucosinolates (Kliebenstein et al., 2001). An addi- gain any insight into the mechanistic links underlying
tional early evidence of the role of specific transcript the observed behavior, given that correlation between
accumulation on a metabolic phenotype came from the biological entities does not always imply causation or
elucidation of the role that different regulation mecha- the existence of functional links (Sweetlove and Fernie,
nisms affecting Trp synthase a and b had on the 2005; Sweetlove et al., 2008; Stitt, 2013). In this regard, it
amount of 2,4-dihydroxy-7-methoxy-1,4-benzoxazin-3- becomes imperative to validate the outputs of coex-
one, a natural pesticide synthesized in maize (Zea mays) pression analyses with follow-up approaches in order
leaves (Melanson et al., 1997). Another example of the to prove the existence of putative functional links. Ar-
coordination between transcripts and metabolite accu- guably, the greatest advances made to date following
mulation came from the analysis of maize anthers, approaches to integrate transcript and metabolite data
where a strong correlation was found between the ex- have been achieved in gene annotation and the struc-
pression of a structural gene (flavanone 3-hydroxylase) tural elucidation of plant intermediary and secondary
and the appearance of specific flavonols (mainly quer- metabolism.
cetin and kaempferol; Deboo et al., 1995). These same Two early studies of particular note are those from
approaches have also been used to select a number of the Saito and Dixon laboratories investigating Arabi-
candidate genes involved in the biosynthesis of cap- dopsis anthocyanin and Medicago truncatula triterpene
saicinoids, a group of vanillylamides conferring pun- metabolism, respectively (Achnine et al., 2005; Tohge
gency to hot peppers (Capsicum spp.). In this case, the et al., 2005). In the case of the anthocyanin pathway,
comparison between sweet and hot pepper varieties prior to the study of Tohge et al. (2005), no late bio-
facilitated the identification of some placenta-specific, synthetic genes involved in anthocyanin decoration
differentially expressed genes that were directly corre- steps had been identified in Arabidopsis, although all
lated with the accumulation of capsaicinoids (Curry early biosynthetic genes have been characterized by
et al., 1999). The examples cited above laid the foun- visible phenotype screening. A combination of tran-
dation for large-scale studies using the parallel analysis script and metabolite profiling on a Production of An-
of transcripts and metabolites. One of the first examples thocyanin Pigment1 activation-tagged line alongside
of this approach focused on the identification of tran- validatory experiments involving both heterologously
scripts strongly correlated with the abundance of given expressed enzymes and knockout mutants resulted in
1500 Plant Physiol. Vol. 169, 2015Integrative Studies of Metabolism
the identification of five genes and the identification of be active under specific conditions. Occasionally,
up to 11 anthocyanins. Such confirmatory experiments however, they can also provide more mechanistic in-
are essential in order to unequivocally assign gene formation. One prominent example of this is the de-
function. The combination of reverse genetic strategies tailed analysis of several transgenic Arabidopsis lines
with the characterization of enzyme activity when the with altered flavonoid levels via transcriptomic and
gene is expressed in a heterologous system remains the metabolomics analyses, including hormone analysis,
gold standard for the molecular identification of novel which revealed that the overaccumulation of fla-
enzyme-catalyzed reactions (Tohge et al., 2005; Luo vonoids exhibiting strong oxidative capacity in vitro
et al., 2007; Yonekura-Sakakibara et al., 2012). Subse- also confers oxidative stress and drought tolerance
quent follow-up studies have identified some six genes (Nakabayashi et al., 2014; Nakabayashi and Saito,
associated with flavonol metabolism, and some 24 2015). In addition, a range of developmental processes
compounds (among 35 compounds found) of this class have been followed at high resolution by a combination
Downloaded from https://academic.oup.com/plphys/article/169/3/1499/6113889 by guest on 28 January 2022
have now been identified in Arabidopsis (Tohge et al., of transcript and metabolite profiles. Such studies are
2007; Yonekura-Sakakibara et al., 2007, 2008, 2014; dominated by studies of fruit ripening (Zamboni et al.,
Nakabayashi et al., 2009; Tohge and Fernie, 2010; Saito 2010; Lin et al., 2015; Vallarino et al., 2015) and leaf
et al., 2013; Fig. 1). While the expansion of the charac- development (Pick et al., 2011; Wang et al., 2014);
terized triterpenoid metabolism in M. truncatula is not however, they are not limited to these processes, with
quite so impressive, the study of Achnine et al. (2005) studies also covering the development of various or-
allowed the functional annotation of 30 different sa- gans, lignin deposition, and the establishment of
ponins, and currently, over 70 metabolites of this com- arbuscular mycorrhizal symbiosis (Vanholme et al.,
pound class have been identified in M. truncatula 2013; Laparre et al., 2014; Nakamura et al., 2014; Wang
(Pollier et al., 2011; Gholami et al., 2014; Watson et al., et al., 2014). In this regard, these approaches prove
2015). The utility of this approach is at its greatest for informative in clarifying the relative importance of
the relatively unchartered pathways of specialized seemingly redundant pathways of biosynthesis and the
metabolism; however, it is worth noting that slight degradation of specific metabolites or may also help
variations on this strategy independently identified the to define the role of those primary metabolites (e.g.
gene encoding plant Thr aldolase (Fernie et al., 2004; g-aminobutyrate) for which a signaling role was hy-
Jander et al., 2004) in Arabidopsis and 2,4-dihydroxy-7- pothesized (Batushansky et al., 2014). For example,
methoxy-1,4-benzoxazin-3-one glucoside methyltrans- ascorbate biosynthesis, which is one of the well-studied
ferase in maize (Meihls et al., 2013). A decade later, the metabolisms in several higher plants, especially in
number of species and pathways for which this ap- Arabidopsis (Wheeler et al., 1998; Gatzek et al., 2002;
proach has been adopted has expanded massively to Laing et al., 2004; Conklin et al., 2006; Dowdle et al.,
include several crops and medicinal plants. Strategies 2007), has been revealed as the dominant route of
combining transcript and metabolite profiling have ascorbate biosynthesis during ripening in tomato
proved effective in elucidating the structure of several (Carrari et al., 2006). Another example could be found in
metabolic pathways involved in the synthesis of pri- the elucidation of the arogenate pathway as an alterna-
mary metabolites, flavonoids, terpenoids, and alkaloids tive route for Phe biosynthesis (Dal Cin et al., 2011). A
(Osorio et al., 2011, 2012; Shelton et al., 2012; Lin et al., similar approach in Arabidopsis, based on feeding
2015). studies and coexpression analysis, allowed an alterna-
On a broader level, the combination of transcript and tive pathway to be proposed for Lys degradation in
metabolite profiling has commonly been used for dark-induced senescent leaves (Araújo et al., 2010).
multilayered descriptions of plant responses, partic- However, despite the fact that these examples illus-
ularly those to abiotic stress (Gibon et al., 2006; trate that combined transcriptome/metabolome stud-
Maruyama-Nakashita et al., 2006; Kusano et al., 2011; ies provide increases in our understanding of the
Gechev et al., 2013; Bielecka et al., 2014; Nakabayashi regulation of metabolic networks, we contend that they
et al., 2014). In this vein, a number of studies have been remain at their most powerful in gene functional an-
carried out that assess the combined transcript and notation and in the elucidation of species- and/or
metabolite responses to water stress, temperature tissue-specific metabolic pathway structures.
stress, light stress, and limitations of nutrient supply
(Urano et al., 2009; Caldana et al., 2011; Kusano et al.,
2011; Nakabayashi et al., 2014). Such studies, while by INTEGRATING METABOLITE AND PROTEOME/
nature descriptive, can afford insight into global met- ENZYME ACTIVITY DATA
abolic variations under certain conditions as well as
identify which pathways are under tight and which are Less commonly used to date than combined tran-
under loose transcriptional control. Given the highly scriptome and metabolome analyses are combined
interconnected nature and nonlinearity of metabolic proteome and metabolome analyses. They are addi-
pathways in the global network structure, and even in tionally largely used in a manner analogous to the more
the absence of flux profiling data, the integration of descriptive studies reviewed above. That said, consid-
transcriptomics with wide metabolic profiling can, in erable insight into metabolic network structure as well
any case, narrow down which metabolic steps could as into general aspects of metabolic regulation have
Plant Physiol. Vol. 169, 2015 1501Tohge et al.
Downloaded from https://academic.oup.com/plphys/article/169/3/1499/6113889 by guest on 28 January 2022
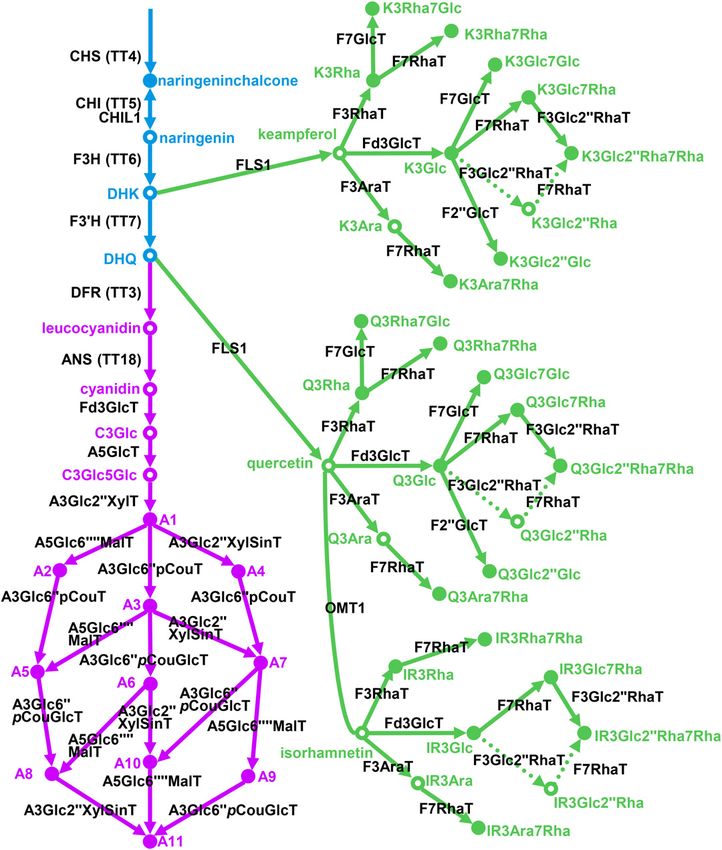
Figure 1. Current model of flavonol/anthocyanin biosynthesis in Arabidopsis. Colors are as follows: blue, early biosynthetic genes;
green, flavonol-specific biosynthetic genes; and purple, anthocyanin-specific biosynthetic genes. CHS, Chalcone synthase, At5g13930; CHI,
chalcone isomerase, At3g55120; CHIL1, At5g05270; F3H, flavanone-3-hydroxylase, At3g51240; F39H, flavonoid 39-hydroxylase, At5g07990;
DFR, dihydroflavonol reductase, At5g42800; ANS, anthocyanidin synthase, At4g22880; F3GlcT, flavonoid-3-O-glucosyltransferase,
UGT78D2, At5g17050; A5GlcT, anthocyanin-5-O-glucosyltransferase, UGT75C1, At4g14090; A3Glc299XylT, anthocyanin-3-O-
glucoside-299-O-xylosyltransferase, UGT79B1, At5g54060; A5Glc69999MalT, anthocyanin-5-O-glucoside-69999-O-malonyltransferase,
At3g29590; A3Glc699pCouT, anthocyanin-3-O-glucoside-699-O-p-coumaroyltransferase, At1g03940, At1g03495; A3Glc299XylSinT,
anthocyanin-3-O-(299-O-xylosyl)-glucoside-6999-O-sinapoyltransferase, At2g23000; A3Glc699pCouT, anthocyanin-3-O-(699-
O-coumaroylglucoside-O-glucosyltransferase, At4g27830; FLS1, flavonol synthase, At5g08640; F3RhaT, flavonol-3-O-rhamnosyltransferase,
UGT78D1, At1g30530; F3AraT, flavonol-3-O-arabinosyltransferase, UGT78D3, At5g17030; F7RhaT, flavonol 7-O-rhamnosyltransferase,
UGT89C1, At1g06000; F7GlcT, flavonol 7-O-glucosyltransferase, UGT73C6, At2g36790; OMT1, O-methyltransferase, At5g54160.
been gained in this manner. Here, we will describe eight parallel to enzyme data (and transcriptomics data)
studies that illustrate how the integration of proteomic across varying diurnal cycles in wild-type and a
and metabolomic data sets has been used to inform starchless mutant of Arabidopsis, revealing that rapid
our understanding of systems regulation. In the first changes in transcripts are integrated over time to gen-
of these examples, metabolite data were studied in erate essentially stable changes in many sectors of
1502 Plant Physiol. Vol. 169, 2015Integrative Studies of Metabolism
metabolism (Gibon et al., 2006). The same group went Tohge et al., 2014); we contend here that an additional
on to apply this approach to tomato fruit development reason to explain this (partial) lack of concordance in
and natural variance in Arabidopsis. In tomato, enzyme the integrative approaches involving metabolism could
profiles were sufficiently characteristic to allow stages lie within the incomplete annotation of most genomes,
of development and cultivars and the wild species to be including those of model organisms. However, we be-
distinguished, but comparison of enzyme activity and lieve the most likely reason to be the lack of linear
metabolites revealed remarkably little connectivity relationship between genes, their protein products, and
between the developmental changes of enzyme and metabolites and, secondly, the fact that most genomes,
metabolite levels, suggesting the operation of post- even those of model organisms, remain incompletely
translational modification mechanisms (Steinhauser annotated. Despite this serious drawback, we hope
et al., 2010). In Arabidopsis, they documented highly to illustrate in this section that the integration of
coordinated changes between enzyme activities, par- metabolomics and genomic data can be incredibly
Downloaded from https://academic.oup.com/plphys/article/169/3/1499/6113889 by guest on 28 January 2022
ticularly within those of the Calvin-Benson cycle, as powerful in understanding natural variation in metab-
well as significant correlations in specific metabolite olism and its regulation.
pairs and between starch and growth. On the other Whole-genome sequences are available for more than
hand, few correlations, and thus low overall connec- 100 plant species (including microalgae; Tohge et al.,
tivity, were observed between enzyme activities and 2014); this massive acceleration afforded by next-
metabolite levels (Sulpice et al., 2010), but strong links generation technologies cannot currently be matched
were seen between starch levels and growth, which we by metabolomics, especially if high-quality species-
describe below. In an alternative approach, proteomic optimized approaches are adopted (Fukushima et al.,
and metabolic data were used merely to extend the 2014). The KNApSAcK database, which is one of the
range of molecular entities in order to demonstrate that largest curated compendia of phytochemicals, contains
fascicular and extrafascicular phloem are isolated from over 700 compounds for early sequenced plants like
one another and divergent in function (Zhang et al., Arabidopsis and rice (Oryza sativa) but no entries for
2010). A similar approach was taken to identify root as recently sequenced species such as goatgrass (Aegilops
the major organ involved in alkaloid biosynthesis in tauschii) and wild tobacco (Nicotiana tomentosiformis).
Macleaya spp. (Zeng et al., 2013). Three further studies In this section, we will describe insight gained from
of note are more similar to that of Gibon et al. (2006) in combining metabolomic data with genome sequences
that they use a combination of proteomics and meta- in three different case studies: (1) a simple comparison
bolomics as a means to define the complex response of of a reference genome with metabolomics data; (2) a
the cell to varying circumstances, be they iron nutrition comparison of natural allelic and metabolic variance;
in Arabidopsis (Sudre et al., 2013), the drought re- and (3) integrating genome sequence data into quanti-
sponse in maize xylem (Alvarez et al., 2008), or heat tative genetics approaches. The first of these has been
stress acclimation in the model alga Chlamydomonas covered in considerable detail recently (Fukushima
reinhardtii (Hemme et al., 2014). The fact that many of et al., 2014; Tohge et al., 2014), so we will only briefly
these studies were published in the last 2 years reflects describe it here. The starting point is to perform
the growing uptake of such strategies. That said, in our genome-wide ortholog searches using functionally an-
opinion, it remains an underexploited research ap- notated genes; best practice is to use cross-species
proach to date. cluster-based BLAST searches such as those housed in
the PLAZA database (Proost et al., 2009) or, in the case of
photosynthetic microbes, pico-PLAZA (Vandepoele et al.,
INTEGRATING METABOLITE AND GENOME DATA
2013). Illustrations of how such analyses have been
performed for central, shikimate, phenylpropanoid,
Given that the advent of metabolomics more or less terpenoid, alkaloid, and glucosinolate metabolism have
paralleled the release of the first plant genome, the been presented (Hofberger et al., 2013; Tohge et al.,
integration of metabolomics and whole-genome se- 2013a, 2013b, 2014; Cavalcanti et al., 2014; Boutanaev
quence data is perhaps unsurprising. The true potential et al., 2015). Thereafter, comparison of these gene in-
of this approach has been realized only within the last ventories with metabolite profiles of the species under
few years; we will not describe it again in detail, given evaluation allows the construction of putative meta-
that it is discussed in a previous correspondence in bolic pathway structures that can be further tested via
Plant Physiology (Fernie and Stitt, 2012). Suffice it to say, reverse genetics or heterologous expression, as de-
there are considerable complexities in such combina- scribed in “Integrating Metabolite and Transcript Data”
tions; tellingly, early studies aimed at computational above. Important insights into pathway evolution can
prediction of the size of the Escherichia coli metabolome be gained from such approaches, as illustrated by the
estimated a complement of approximately 750 metab- recent cross-kingdom comparison of ascorbate biosyn-
olites, while subsequent experimental approaches have thesis (Wheeler et al., 2015).
revealed many metabolites that were not computed The second case study, that of evaluating allelic and
from the genome (van der Werf et al., 2007). Several metabolic variance across natural diversity, is similar in
potential reasons could be put forward to explain this scope yet far more targeted than genome-wide associ-
discrepancy (for review, see Fernie and Stitt, 2012; ation studies, which we describe below. The majority of
Plant Physiol. Vol. 169, 2015 1503Tohge et al.
recent examples of its utility come from the analysis of availability of the sequences of both parental genomes
wild species tomato; however, it is important to note (Bolger et al., 2014) narrowed down the origin of the
that the approach itself is essentially just a modification metabolic variation to specific genetic polymorphisms
of that adopted over decades in the cloning of natural in some selected metabolic quantitative trait loci
color mutants (Fernie and Klee, 2011). In the last few (Quadrana et al., 2014; Alseekh et al., 2015). The inte-
years, understanding of primary as well as secondary gration between genotypic and metabolic variance can
and cuticular cell wall metabolism has been enhanced be, and has actually been applied, also on large col-
considerably via this approach (Schauer et al., 2005; lections of unrelated individuals (metabolite-based
Matas et al., 2011; Kim et al., 2012, 2014; Koenig et al., genome-wide association studies): as in the case of
2013), albeit the greatest insight into the latter was biparental populations, also with this strategy, several
ultimately elucidated via the use of an introgression line cases of polymorphological variants of genomic se-
population, as described below. In essence, this ap- quences have been identified and related to metabolic
Downloaded from https://academic.oup.com/plphys/article/169/3/1499/6113889 by guest on 28 January 2022
proach starts with the identification of metabolic vari- variation. These two approaches, based either on bi-
ance within a population of ecotypes, cultivars, or parental populations or on large collections of natural
similarly related species and attempts to link this with accessions, have been used in Arabidopsis and crop
allelic diversity or gene duplication, as has been species (maize, rice, wheat [Triticum aestivum], and fruit
achieved for acyl-sugar metabolites (Schilmiller et al., trees; Gong et al., 2013; Li et al., 2013; Wen et al., 2014;
2015), terpenes (Matsuba et al., 2013), and isoprenoids Matsuda et al., 2015; for review, see Luo, 2015). The
(Kang et al., 2014), or even with the presence or absence boon that new sequences will provide, especially from
of genes, as described recently for methylated flavo- wild relatives or locally adapted varieties, will be rep-
noids of glandular trichomes (Kim et al., 2014). The resented by the possibility to dissect the genetic basis of
preceding list documents the success of this approach; metabolite variation, with a view to introgress benefi-
until recently, however, it was constrained by the limits cial traits in crop improvement.
of our a priori knowledge, which is needed in order to
select the candidate genes in which we search for allelic
variance. The development of RNA sequencing tech-
INTEGRATING METABOLITE AND
nologies means that we are no longer limited by the
PHYSIOLOGICAL DATA
amount of sequence data; a potential hurdle to these
integrative approaches, however, can still be present While the above examples concentrate on the inte-
when comparing highly genetically divergent individ- gration of various types of profiling data with one
uals, since the number of genetic polymorphisms is another in order to advance our understanding of
too great to evaluate one by one. For this reason, the metabolic pathway structure and/or metabolic regu-
quantitative trait loci approach is a powerful alternative lation, relatively few studies have attempted to cor-
method of associating phenotypes to their underlying relate metabolite content with physiological data,
genetic variance. The use of such approaches in plant including growth and yield (for review, see Stitt et al.,
metabolism has been the subject of several recent 2010; Carreno-Quintero et al., 2013). One of the earliest
comprehensive reviews (Kliebenstein, 2009; Scossa studies to do so was the above-described metabolic
et al., 2015); however, we will provide a couple of quantitative trait loci analysis of the S. pennellii intro-
examples of their utility for advancing the under- gression lines, in which yield-associated plant traits
standing of metabolite accumulation and metabolic were measured alongside primary metabolite content
regulation. of the fruit (Schauer et al., 2006). In this study, network
The fruit of tomato, as the model species for ripen- analysis based on cartographic modeling algorithms
ing of fleshy fruits, has been the subject of combined developed by Guimerà and Nunes Amaral (2005)
large-scale genomic, physiological, and metabolic in- identified that yield-associated traits were positively
vestigations, often making use of specific biparental correlated to a range of previously defined signal me-
populations or large sets of unrelated individuals, in tabolites, compounds that have signaling as well as
an attempt to understand the causal variants of the metabolic functions, including Suc, hexose, and inositol
metabolic variations (Schauer et al., 2005; Lin et al., phosphates, Pro, and g-aminobutyrate. In addition, this
2014; Sauvage et al., 2014). In particular, the use of a study indicated that the harvest index (i.e. the ratio of
population of introgression lines, obtained from the harvestable product to total biomass) negatively cor-
cross between tomato and Solanum pennellii (a wild related with the content of the vast majority of amino
tomato species), has greatly aided the identification of acids. This relationship was confirmed in an indepen-
quantitative trait loci for a large number of physio- dent population and following experiments that artifi-
logical and metabolic traits. Profiling data of primary cially altered the fruit load per truss (Do et al., 2010).
and secondary metabolites in this population were However, as would perhaps be anticipated, subsequent
collected over several years (along with some classi- evaluation of the relationship between growth and
cal yield-related traits), revealing more than 1,500 secondary metabolite content revealed far less correla-
metabolic quantitative trait loci affecting the levels tion (Alseekh et al., 2015). Using essentially the same
of several sugars, amino acids, organic acids, vita- approach in an Arabidopsis recombinant inbred line
mins, phenylpropanoids, and glycoalkaloids. The population, Meyer et al. (2007) found that, although no
1504 Plant Physiol. Vol. 169, 2015Integrative Studies of Metabolism
single metabolite exhibited a very high correlation with natural environments, the accumulation of capsaicinoids
biomass, canonical correlation analysis in which the in populations of Capsicum chacoense (a wild pepper
data of a linear combination of metabolites allowed the species) is inversely correlated with seed set; these
improvement of this correlation by a factor of 10, thus metabolites, however, have a defensive role in highly
defined a metabolic signature of growth. Intriguingly, humid environments, where their accumulation deters
the hexose phosphates Glc-6-P and Fru-6-P as well as the attack of phytopathogenic fungi. Across a geo-
Suc were among the 20 top metabolites contributing graphical gradient of decreasing rainfall (with a grad-
to this signature. When similar approaches were ap- ual decreasing pressure of the pathogens, which thrive
plied to maize, strong genome-wide association links only in humid environments), the accumulation of
were found between coumaric and caffeic acids and capsaicinoids also decreases in Capsicum spp. popula-
cinnamoyl-CoA reductase, while these precursors also tions, while seed set, on the other hand, increases. This
significantly correlated with lignin content plant height study is an example of the combination of targeted
Downloaded from https://academic.oup.com/plphys/article/169/3/1499/6113889 by guest on 28 January 2022
and dry matter yield, presenting another example of the metabolic approaches with population ecology in dis-
narrowing of the genotype-phenotype gap of complex secting the basis of natural polymorphic traits (Haak
agronomic traits (Riedelsheimer et al., 2012). The same et al., 2012). Further studies that address the concept of
group revealed that models based on data obtained for metabolism and growth tradeoffs have used reciprocal
130 metabolites gave highly accurate predictions of crosses to assess the contribution of the organellar ge-
agronomic traits and suggested that combined metab- nome to the processes and came to the conclusion that
olite, genomic, and agronomic phenotyping represents there is far greater diversity in defense chemistry than
an important screening tool for the identification of primary metabolism (Joseph et al., 2013, 2015). The in-
parental lines for the creation of superior hybrid crops terrogation of such tradeoffs is only possible via the
(Riedelsheimer et al., 2012). integrated approach described here and appears to be
Returning to Arabidopsis, evaluation of the variation very powerful; as such, we would expect considerable
of growth, metabolite levels, and enzyme activities was advances in our understanding of this phenomenon to
also carried out across 94 accessions, revealing that be gained following its application. Not just the last
biomass correlated negatively with many metabolites, three studies but all of the above studies have been
including starch and protein and to a much lesser extent published within the last 6 years, reflecting the fact
Suc (Sulpice et al., 2009). However, further experiments that such analyses are in their infancy. Given the
in which 97 accessions grown in near-optimal carbon recognized complexity of the metabolism-to-growth
and nitrogen supply, restricted carbon supply, and re- interactions, a comprehensive understanding of the
stricted nitrogen supply and analyzed for biomass and intricate networks that coordinate this interface is
54 metabolic traits revealed that robust prediction of likely some time off. That said, as the above examples
phenotypic traits (biomass, starch, and protein) is most illustrate, the integration of growth data into metab-
effective (and reliable) when metabolite data (upon olite profiling data as well as that of simpler physio-
which predictions are based) are collected from the logical processes such as photosynthetic or respiratory
same growth environments (Meyer et al., 2007; Sulpice rates (Florez-Sarasa et al., 2012) has already presented
et al., 2009; Korn et al., 2010; Steinfath et al., 2010). a number of key findings.
Clearly, attempting to predict biomass, for example,
from metabolic profiles collected in a different growth
environment generally yields fewer (and weaker) cor- POSTGENOMIC INTEGRATION OF DATABASE-
relations (Sulpice et al., 2013). Therefore, the prediction HOUSED RESEARCH WITH NOVEL EXPERIMENTS
of biomass across a range of conditions would bet-
ter require condition-specific measurement of meta- The examples described above rely on the integration
bolic traits to take account of environment-dependent of data obtained in parallel using different experimen-
changes of the underlying networks (Sulpice et al., tal approaches. While such approaches are ideal for
2013). Data from this study were subsequently ana- addressing a number of questions, particularly those
lyzed with respect to the tradeoffs between metabolism concerning the temporal aspects underlying dynamic
and growth, specifically comparing increasing size with responses to a systems perturbation, the integration
increasing protein concentration, demonstrating that of novel experimental data with different types of
accessions with high metabolic efficiency lie closer to archived data can also prove highly informative, pro-
the Pareto performance frontier (the optimal solution viding an appropriate amount of caution is used in
for the two contending tasks) and hence exhibit in- interpreting the results. Here, we will provide several
creased metabolic plasticity (Kleessen et al., 2014). A examples illustrative of such approaches, which largely
related study addressing an ecological tradeoff between fit into two major types of approaches: (1) those using
secondary metabolism and fitness relates to the accu- correlative approaches and (2) those using genome-
mulation of capsaicinoids in the placenta of pepper scale stoichiometric models. The first study we will
fruits (Capsicum spp.). Capsaicinoids constitute a class describe fits into the former category, being an attempt
of vanillylamides derived from Phe; they accumulate to define the storage metabolome of the vacuole (Tohge
in ripening pepper fruit and are responsible for the et al., 2011; Fig. 2). In this research, a combination of
pungency sensation occurring upon ingestion. In gas chromatography-mass spectrometry and Fourier
Plant Physiol. Vol. 169, 2015 1505Tohge et al.
previously described transporter proteins as well as to
highlight the dynamic nature of the storage metabolome.
The coexpression approach has also been combined
with metabolic profiling in the annotation of plasma
membrane lignin and plastidial glycolate/glycerate
and bile acid transporters (Gigolashvili et al., 2009;
Sawada et al., 2009; Alejandro et al., 2012) as well as
a multitude of cell wall-associated proteins (Persson
et al., 2005). Moreover, this approach has also been used
to identify process, as opposed to pathway-specific, pro-
teins, identifying proteins involved in dark-induced
senescence (Araújo et al., 2011) and in the response to
Downloaded from https://academic.oup.com/plphys/article/169/3/1499/6113889 by guest on 28 January 2022
UV-B irradiance (Kusano et al., 2011).
The other type of examples we would like to discuss
are based on the integration of transcriptomic and
metabolomics level genome-scale models (Töpfer et al.,
2014). In the first of these studies, microarray data from
Arabidopsis exposed to eight different light and tem-
perature conditions (data published in Caldana et al.,
2011) were integrated into a genome-scale model
(Mintz-Oron et al., 2012). Before discussing the out-
come of this integration, we first digress to provide a
brief description of how genome-scale models are
Figure 2. Schematic overview of an integrative approach using me- generated. Essentially, a genome-scale model corre-
tabolite profiling of storage metabolite and membrane proteomic data. sponds metabolic genes with metabolic pathways in a
Example of network: barley vacuole network from Tohge et al. (2011).
manner whereby a stoichiometrically balanced meta-
bolic network is generated, which corresponds to all
gene functions annotated for that organism. Such
transform mass spectrometry was used to detect and models were originally published for microbes at the
quantify some 59 primary metabolites and 200 sec- turn of the century (Edwards and Palsson, 2000), with
ondary metabolites (defined on the basis of strong many models for plants species being subsequently
chemical formulae predictions) in either silicon oil- generated, including the model species Arabidopsis as
purified barley (Hordeum vulgare) vacuoles or the pro- well as crop species such as rice and maize (for review,
toplasts from which these were derived. Of the 259 see Simons et al., 2014). Returning to the superimposi-
putative metabolites, 12 were exclusively detected in tion of experimental data on the model, the addition of
the vacuole, 34 were exclusively in the protoplast, and transcriptomic data was able to predict flux capacities
213 were common to both samples. At the quantitative and statistically assess whether these vary under the
level, the difference between vacuole and protoplast experimental conditions tested. Moreover, this study
was yet more striking, with secondary metabolites be- introduced the concepts of metabolic sustainers and
ing differentially abundant between the two sample modulators, with the former being metabolic functions
types. As a next step to predict the underlying cytosolic- that are differentially up-regulated with respect to the
vacuolar transporters, tonoplast proteins predicted to null model whereas the latter are differentially down-
have a transport function were evaluated within the regulated in order to control a certain flux and, there-
context of the metabolic profiling data. Specifically, 88 fore, modulate affected processes (Töpfer et al., 2013).
proteins reported to be tonoplast proteins in barley In a follow-up study, predictions made from the inte-
(Endler et al., 2006) were evaluated after conversion to gration of transcriptomics were complemented with
Affymetrix probe identifiers and coexpression analysis metabolomics data from the same experiment. In doing
of the resultant 128 probe sets was carried out using so, the authors were able to bridge flux-centric and
PlaNet for barley (Mutwil et al., 2011; http://aranet. metabolomics-centric approaches and, in so doing,
mpimp-golm.mpg.de). Coexpressed networks of these demonstrate that, under certain conditions, metabolites
probes separated into 13 subgroups, with the most serving as pathway substrates in pathways defined as
dense cluster being highly correlated with aromatic either modulators or sustainers display lower temporal
amino acid-related genes and the second most dense variation with respect to all other metabolites (Töpfer
cluster including several vacuolar ATP synthase pro- et al., 2013). These findings are thus in concordance
teins and tricarboxylic acid cycle-related genes. In with theories of network rigidity and pathway robust-
addition, clear associations were found between the ness (Stephanopoulos and Vallino, 1991; Rontein et al.,
expression of transport proteins and that of pathways 2002; Williams et al., 2008). Furthermore, considerable
of flavonoid and mugineic acid synthesis as well evidence suggests that the levels of specific metabolites,
as storage protein functions (Tohge et al., 2011). This such as Ala, pyruvate, 2-oxoglutarate, Gln, and sper-
study was thus able to putatively assign function to midine, are exceptionally stable across a massive range
1506 Plant Physiol. Vol. 169, 2015Integrative Studies of Metabolism
of cellular circumstances (Geigenberger, 2003; Stitt Alejandro S, Lee Y, Tohge T, Sudre D, Osorio S, Park J, Bovet L, Lee Y,
Geldner N, Fernie AR, et al (2012) AtABCG29 is a monolignol trans-
and Fernie, 2003). They also are in keeping with porter involved in lignin biosynthesis. Curr Biol 22: 1207–1212
observations that the levels of metabolites such as Ser Alseekh S, Tohge T, Wendenberg R, Scossa F, Omranian N, Li J, Kleessen
coordinately control the levels of expression of genes S, Giavalisco P, Pleban T, Mueller-Roeber B, et al (2015) Identification
encoding multiple steps of the pathways to which they, and mode of inheritance of quantitative trait loci for secondary metab-
themselves, belong (Timm et al., 2013). The high sta- olite abundance in tomato. Plant Cell 27: 485–512
Alvarez S, Marsh EL, Schroeder SG, Schachtman DP (2008) Metabolomic
bility of these metabolites is in keeping with their re- and proteomic changes in the xylem sap of maize under drought. Plant
quirement across a range of different stresses. It also Cell Environ 31: 325–340
highlights the fact that the robust metabolites may well Araújo WL, Ishizaki K, Nunes-Nesi A, Larson TR, Tohge T, Krahnert I,
be the most biologically relevant for metabolic regula- Witt S, Obata T, Schauer N, Graham IA, et al (2010) Identification of
the 2-hydroxyglutarate and isovaleryl-CoA dehydrogenases as alterna-
tion; this is an important point, since it is at odds with tive electron donors linking lysine catabolism to the electron transport
the manner in which the majority of the metabolomics chain of Arabidopsis mitochondria. Plant Cell 22: 1549–1563
Downloaded from https://academic.oup.com/plphys/article/169/3/1499/6113889 by guest on 28 January 2022
community assesses their data. This observation addi- Araújo WL, Ishizaki K, Nunes-Nesi A, Tohge T, Larson TR, Krahnert I,
tionally highlights the potential difficulties and chal- Balbo I, Witt S, Dörmann P, Graham IA, et al (2011) Analysis of a range
lenges in interpreting data from a single level of the of catabolic mutants provides evidence that phytanoyl-coenzyme A
does not act as a substrate of the electron-transfer flavoprotein/electron-
cellular hierarchy and thus provides further grounds transfer flavoprotein:ubiquinone oxidoreductase complex in Arabidopsis
for integrated models. during dark-induced senescence. Plant Physiol 157: 55–69
Bartling D, Seedorf M, Schmidt RC, Weiler EW (1994) Molecular char-
acterization of two cloned nitrilases from Arabidopsis thaliana: key
enzymes in biosynthesis of the plant hormone indole-3-acetic acid. Proc
CURRENT AND FUTURE CHALLENGES IN
Natl Acad Sci USA 91: 6021–6025
DATA INTEGRATION Batushansky A, Kirma M, Grillich N, Toubiana D, Pham PA, Balbo I,
Fromm H, Galili G, Fernie AR, Fait A (2014) Combined transcriptomics
The above sections document that integrative ap- and metabolomics of Arabidopsis thaliana seedlings exposed to exoge-
proaches to further our understanding of metabolism nous GABA suggest its role in plants is predominantly metabolic. Mol
have proven very successful over the last decade or so, Plant 7: 1065–1068
particularly when linked with genetic and/or envi- Bielecka M, Watanabe M, Morcuende R, Scheible WR, Hawkesford MJ,
Hesse H, Hoefgen R (2014) Transcriptome and metabolome analysis of
ronmental experiments. To date, the approaches taken plant sulfate starvation and resupply provides novel information on
have been relatively straightforward and have gener- transcriptional regulation of metabolism associated with sulfur, nitro-
ally not been performed at a high level of spatial reso- gen and phosphorus nutritional responses in Arabidopsis. Front Plant
lution. Several methods currently exist to obtain data Sci 5: 805
from all of the methods described here at the tissue, Bolger A, Scossa F, Bolger ME, Lanz C, Maumus F, Tohge T, Quesneville
H, Alseekh S, Sørensen I, Lichtenstein G, et al (2014) The genome of
cellular, and even subcellular levels (Aharoni and the stress-tolerant wild tomato species Solanum pennellii. Nat Genet 46:
Brandizzi, 2012); while still technically challenging, it 1034–1038
seems conceivable that such methods could provide Boutanaev AM, Moses T, Zi J, Nelson DR, Mugford ST, Peters RJ,
data required to better understand the cell specializa- Osbourn A (2015) Investigation of terpene diversification across mul-
tiple sequenced plant genomes. Proc Natl Acad Sci USA 112: E81–E88
tion of metabolism. In addition, methods to gain accu- Caldana C, Degenkolbe T, Cuadros-Inostroza A, Klie S, Sulpice R, Leisse
rate metabolic flux estimates following 13CO2 labeling A, Steinhauser D, Fernie AR, Willmitzer L, Hannah MA (2011) High-
have recently been established (Young et al., 2008; density kinetic analysis of the metabolomic and transcriptomic response
Szecowka et al., 2013; Ma et al., 2014) but are not yet of Arabidopsis to eight environmental conditions. Plant J 67: 869–884
fully integrated with protein or transcript data. How- Carrari F, Baxter C, Usadel B, Urbanczyk-Wochniak E, Zanor MI, Nunes-
Nesi A, Nikiforova V, Centero D, Ratzka A, Pauly M, et al (2006)
ever, it is important to note that such experiments, al- Integrated analysis of metabolite and transcript levels reveals the
beit using [13C]Glc as a precursor, have already been metabolic shifts that underlie tomato fruit development and highlight
carried out in in vitro-cultivated Brassica napus em- regulatory aspects of metabolic network behavior. Plant Physiol 142:
bryos, providing considerable insight into the systems- 1380–1396
Carreno-Quintero N, Bouwmeester HJ, Keurentjes JJB (2013) Genetic
level regulation of this organ (Schwender et al., 2015). It analysis of metabolome-phenotype interactions: from model to crop
additionally seems highly likely that future research species. Trends Genet 29: 41–50
will draw more heavily on archived genomics data than Cavalcanti JH, Esteves-Ferreira AA, Quinhones CG, Pereira-Lima IA,
it has to date; thus, the continued availability and Nunes-Nesi A, Fernie AR, Araújo WL (2014) Evolution and functional
quality-control curation of such data sets are imperative implications of the tricarboxylic acid cycle as revealed by phylogenetic
analysis. Genome Biol Evol 6: 2830–2848
if we are going to fully exploit their value. Centeno DC, Osorio S, Nunes-Nesi A, Bertolo ALF, Carneiro RT, Araújo
Received July 7, 2015; accepted September 10, 2015; published September 14, WL, Steinhauser MC, Michalska J, Rohrmann J, Geigenberger P, et al
2015. (2011) Malate plays a crucial role in starch metabolism, ripening, and
soluble solid content of tomato fruit and affects postharvest softening.
Plant Cell 23: 162–184
LITERATURE CITED Conklin PL, Gatzek S, Wheeler GL, Dowdle J, Raymond MJ, Rolinski S,
Isupov M, Littlechild JA, Smirnoff N (2006) Arabidopsis thaliana VTC4
Achnine L, Huhman DV, Farag MA, Sumner LW, Blount JW, Dixon RA encodes L-galactose-1-P phosphatase, a plant ascorbic acid biosynthetic
(2005) Genomics-based selection and functional characterization of enzyme. J Biol Chem 281: 15662–15670
triterpene glycosyltransferases from the model legume Medicago Curry J, Aluru M, Mendoza M, Nevarez J, Melendrez M, O’Connell MA
truncatula. Plant J 41: 875–887 (1999) Transcripts for possible capsaicinoid biosynthetic genes are dif-
Aharoni A, Brandizzi F (2012) High-resolution measurements in plant bi- ferentially accumulated in pungent and non-pungent Capsicum spp.
ology. Plant J 70: 1–4 Plant Sci 148: 47–57
Plant Physiol. Vol. 169, 2015 1507Tohge et al.
Dal Cin V, Tieman DM, Tohge T, McQuinn R, de Vos RCH, Osorio S, Góngora-Castillo E, Childs KL, Fedewa G, Hamilton JP, Liscombe DK,
Schmelz EA, Taylor MG, Smits-Kroon MT, Schuurink RC, et al (2011) Magallanes-Lundback M, Mandadi KK, Nims E, Runguphan W,
Identification of genes in the phenylalanine metabolic pathway by ec- Vaillancourt B, et al (2012) Development of transcriptomic resources for
topic expression of a MYB transcription factor in tomato fruit. Plant Cell interrogating the biosynthesis of monoterpene indole alkaloids in me-
23: 2738–2753 dicinal plant species. PLoS One 7: e52506
Deboo GB, Albertsen MC, Taylor LP (1995) Flavanone 3-hydroxylase Guimerà R, Nunes Amaral LA (2005) Functional cartography of complex
transcripts and flavonol accumulation are temporally coordinate in metabolic networks. Nature 433: 895–900
maize anthers. Plant J 7: 703–713 Haak DC, McGinnis LA, Levey DJ, Tewksbury JJ (2012) Why are not all
Dixon RA, Strack D (2003) Phytochemistry meets genome analysis, and chilies hot? A trade-off limits pungency. Proc Biol Sci 279: 2012–2017
beyond. Phytochemistry 62: 815–816 Hannah MA, Caldana C, Steinhauser D, Balbo I, Fernie AR, Willmitzer L
Do PT, Prudent M, Sulpice R, Causse M, Fernie AR (2010) The influence of (2010) Combined transcript and metabolite profiling of Arabidopsis
fruit load on the tomato pericarp metabolome in a Solanum chmielewskii grown under widely variant growth conditions facilitates the identifi-
introgression line population. Plant Physiol 154: 1128–1142 cation of novel metabolite-mediated regulation of gene expression. Plant
Dowdle J, Ishikawa T, Gatzek S, Rolinski S, Smirnoff N (2007) Two genes Physiol 152: 2120–2129
in Arabidopsis thaliana encoding GDP-L-galactose phosphorylase are Hemme D, Veyel D, Mühlhaus T, Sommer F, Jüppner J, Unger AK,
Downloaded from https://academic.oup.com/plphys/article/169/3/1499/6113889 by guest on 28 January 2022
required for ascorbate biosynthesis and seedling viability. Plant J 52: Sandmann M, Fehrle I, Schönfelder S, Steup M, et al (2014) Systems-
673–689 wide analysis of acclimation responses to long-term heat stress and re-
Eastmond PJ, Astley HM, Parsley K, Aubry S, Williams BP, Menard GN, covery in the photosynthetic model organism Chlamydomonas reinhardtii.
Craddock CP, Nunes-Nesi A, Fernie AR, Hibberd JM (2015) Arabi- Plant Cell 26: 4270–4297
dopsis uses two gluconeogenic gateways for organic acids to fuel Hill AT, Ying S, Plaxton WC (2014) Phosphorylation of bacterial-type
seedling establishment. Nat Commun 6: 6659 phosphoenolpyruvate carboxylase by a Ca2+-dependent protein kinase
Edwards JS, Palsson BO (2000) The Escherichia coli MG1655 in silico suggests a link between Ca2+ signalling and anaplerotic pathway control
metabolic genotype: its definition, characteristics, and capabilities. Proc in developing castor oil seeds. Biochem J 458: 109–118
Natl Acad Sci USA 97: 5528–5533 Hirai MY, Sugiyama K, Sawada Y, Tohge T, Obayashi T, Suzuki A, Araki
Endler A, Meyer S, Schelbert S, Schneider T, Weschke W, Peters SW, R, Sakurai N, Suzuki H, Aoki K, et al (2007) Omics-based identification
Keller F, Baginsky S, Martinoia E, Schmidt UG (2006) Identification of of Arabidopsis Myb transcription factors regulating aliphatic gluco-
a vacuolar sucrose transporter in barley and Arabidopsis mesophyll sinolate biosynthesis. Proc Natl Acad Sci USA 104: 6478–6483
cells by a tonoplast proteomic approach. Plant Physiol 141: 196–207 Hirai MY, Yano M, Goodenowe DB, Kanaya S, Kimura T, Awazuhara M,
Fernie AR (2007) The future of metabolic phytochemistry: larger numbers Arita M, Fujiwara T, Saito K (2004) Integration of transcriptomics and
of metabolites, higher resolution, greater understanding. Phytochemis- metabolomics for understanding of global responses to nutritional
try 68: 2861–2880 stresses in Arabidopsis thaliana. Proc Natl Acad Sci USA 101: 10205–
Fernie AR, Klee HJ (2011) The use of natural genetic diversity in the un- 10210
derstanding of metabolic organization and regulation. Front Plant Sci 2: Hofberger JA, Lyons E, Edger PP, Pires JC, Schranz ME (2013) Whole
59 genome and tandem duplicate retention facilitated glucosinolate path-
Fernie AR, Stitt M (2012) On the discordance of metabolomics with pro- way diversification in the mustard family. Genome Biol Evol 5: 2155–
teomics and transcriptomics: coping with increasing complexity in logic, 2173
chemistry, and network interactions. Plant Physiol 158: 1139–1145 Jander G, Norris SR, Joshi V, Fraga M, Rugg A, Yu S, Li L, Last RL (2004)
Fernie AR, Trethewey RN, Krotzky AJ, Willmitzer L (2004) Metabolite Application of a high-throughput HPLC-MS/MS assay to Arabidopsis
profiling: from diagnostics to systems biology. Nat Rev Mol Cell Biol 5: mutant screening; evidence that threonine aldolase plays a role in seed
763–769 nutritional quality. Plant J 39: 465–475
Florez-Sarasa I, Araújo WL, Wallström SV, Rasmusson AG, Fernie AR, Joseph B, Corwin JA, Kliebenstein DJ (2015) Genetic variation in the
Ribas-Carbo M (2012) Light-responsive metabolite and transcript levels nuclear and organellar genomes modulates stochastic variation in the
are maintained following a dark-adaptation period in leaves of Arabi- metabolome, growth, and defense. PLoS Genet 11: e1004779
dopsis thaliana. New Phytol 195: 136–148 Joseph B, Corwin JA, Züst T, Li B, Iravani M, Schaepman-Strub G,
Fukushima A, Kanaya S, Nishida K (2014) Integrated network analysis Turnbull LA, Kliebenstein DJ (2013) Hierarchical nuclear and cyto-
and effective tools in plant systems biology. Front Plant Sci 5: 598 plasmic genetic architectures for plant growth and defense within Ara-
Futuyma DJ, Agrawal AA (2009) Macroevolution and the biological di- bidopsis. Plant Cell 25: 1929–1945
versity of plants and herbivores. Proc Natl Acad Sci USA 106: 18054– Kang JH, Gonzales-Vigil E, Matsuba Y, Pichersky E, Barry CS (2014)
18061 Determination of residues responsible for substrate and product speci-
Gatzek S, Wheeler GL, Smirnoff N (2002) Antisense suppression of ficity of Solanum habrochaites short-chain cis-prenyltransferases. Plant
L-galactose dehydrogenase in Arabidopsis thaliana provides evidence Physiol 164: 80–91
for its role in ascorbate synthesis and reveals light modulated Kim J, Kang K, Gonzales-Vigil E, Shi F, Jones AD, Barry CS, Last RL
L-galactose synthesis. Plant J 30: 541–553 (2012) Striking natural diversity in glandular trichome acylsugar com-
Gechev TS, Benina M, Obata T, Tohge T, Sujeeth N, Minkov I, Hille J, position is shaped by variation at the Acyltransferase2 locus in the wild
Temanni MR, Marriott AS, Bergström E, et al (2013) Molecular tomato Solanum habrochaites. Plant Physiol 160: 1854–1870
mechanisms of desiccation tolerance in the resurrection glacial relic Kim J, Matsuba Y, Ning J, Schilmiller AL, Hammar D, Jones AD,
Haberlea rhodopensis. Cell Mol Life Sci 70: 689–709 Pichersky E, Last RL (2014) Analysis of natural and induced variation in
Geigenberger P (2003) Response of plant metabolism to too little oxygen. tomato glandular trichome flavonoids identifies a gene not present in
Curr Opin Plant Biol 6: 247–256 the reference genome. Plant Cell 26: 3272–3285
Gholami A, De Geyter N, Pollier J, Goormachtig S, Goossens A (2014) Kleessen S, Laitinen R, Fusari CM, Antonio C, Sulpice R, Fernie AR, Stitt
Natural product biosynthesis in Medicago species. Nat Prod Rep 31: M, Nikoloski Z (2014) Metabolic efficiency underpins performance
356–380 trade-offs in growth of Arabidopsis thaliana. Nat Commun 5: 3537
Gibon Y, Usadel B, Blaesing OE, Kamlage B, Hoehne M, Trethewey R, Kliebenstein D (2009) Advancing genetic theory and application by met-
Stitt M (2006) Integration of metabolite with transcript and enzyme abolic quantitative trait loci analysis. Plant Cell 21: 1637–1646
activity profiling during diurnal cycles in Arabidopsis rosettes. Genome Kliebenstein DJ, Lambrix VM, Reichelt M, Gershenzon J, Mitchell-Olds
Biol 7: R76 T (2001) Gene duplication in the diversification of secondary metabo-
Gigolashvili T, Yatusevich R, Rollwitz I, Humphry M, Gershenzon J, lism: tandem 2-oxoglutarate-dependent dioxygenases control gluco-
Flügge UI (2009) The plastidic bile acid transporter 5 is required for sinolate biosynthesis in Arabidopsis. Plant Cell 13: 681–693
the biosynthesis of methionine-derived glucosinolates in Arabidopsis Koenig D, Jiménez-Gómez JM, Kimura S, Fulop D, Chitwood DH,
thaliana. Plant Cell 21: 1813–1829 Headland LR, Kumar R, Covington MF, Devisetty UK, Tat AV, et al
Gong L, Chen W, Gao Y, Liu X, Zhang H, Xu C, Yu S, Zhang Q, Luo J (2013) Comparative transcriptomics reveals patterns of selection in
(2013) Genetic analysis of the metabolome exemplified using a rice domesticated and wild tomato. Proc Natl Acad Sci USA 110: E2655–
population. Proc Natl Acad Sci USA 110: 20320–20325 E2662
1508 Plant Physiol. Vol. 169, 2015You can also read

















































